A Practical Guide to Developing Accurate UV-Vis Calibration Curves for Compound Quantification in Biomedical Research
This article provides a comprehensive guide for researchers and drug development professionals on developing and validating UV-Vis calibration curves for precise compound quantification.
A Practical Guide to Developing Accurate UV-Vis Calibration Curves for Compound Quantification in Biomedical Research
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on developing and validating UV-Vis calibration curves for precise compound quantification. It covers foundational principles grounded in the Beer-Lambert Law, detailed methodological protocols for creating linear and non-linear curves, advanced troubleshooting for common instrument and sample issues, and a comparative analysis of UV-Vis against other quantification techniques like HPLC and NMR. By integrating foundational knowledge with practical application, troubleshooting, and validation strategies, this resource aims to enhance data reliability and methodological robustness in pharmaceutical and clinical research settings.
UV-Vis Spectroscopy and Calibration Fundamentals: Principles for Accurate Quantification
The Beer-Lambert Law (also known as Beer's Law) is a fundamental principle in spectroscopy that establishes a linear relationship between the absorbance of light by a substance and its concentration [1]. This law serves as the cornerstone for quantitative analysis in ultraviolet-visible (UV-Vis) spectroscopy, enabling researchers to determine the concentration of analytes in solution by measuring how much light they absorb [2] [3]. In the context of drug development and analytical research, this principle provides the theoretical foundation for developing robust calibration curves essential for accurate compound quantification [4].
The law is mathematically expressed as: A = εbc Where:
- A is the measured absorbance (a dimensionless quantity)
- ε (epsilon) is the molar absorptivity or molar extinction coefficient (L·mol⁻¹·cm⁻¹)
- b is the path length of light through the solution (cm)
- c is the concentration of the absorbing species (mol·L⁻¹) [1] [3]
This relationship indicates that absorbance is directly proportional to both the concentration of the substance and the path length of the light through the sample, with the molar absorptivity representing how strongly a chemical species absorbs light at a specific wavelength [5].
Theoretical Foundation
Fundamental Concepts of Light Absorption
When monochromatic light passes through a solution containing an absorbing species, photons interact with molecules, promoting electrons to higher energy states. This interaction results in a measurable attenuation of the incident light beam [2]. The extent of light absorption depends on several factors, including the molecular structure of the analyte, the wavelength of light used, and the number of molecules in the light path [1].
The relationship between incident and transmitted light intensity is described through two key parameters:
- Transmittance (T): The ratio of transmitted light intensity (I) to incident light intensity (I₀), often expressed as a percentage [1]
- Absorbance (A): The logarithmic measure of light attenuation by the sample, calculated as A = log₁₀(I₀/I) [1] [3]
The following table illustrates the inverse logarithmic relationship between absorbance and transmittance:
| Absorbance (A) | Transmittance (T) |
|---|---|
| 0 | 100% |
| 0.3 | 50% |
| 1 | 10% |
| 2 | 1% |
| 3 | 0.1% |
| 4 | 0.01% |
Table 1: Relationship between absorbance and transmittance values [1]
The Components of the Beer-Lambert Equation
Molar Absorptivity (ε) The molar absorptivity coefficient is a substance-specific constant that measures how effectively a chemical species absorbs light at a particular wavelength [3]. This intrinsic molecular property depends on the electronic structure of the molecule and the solvent system used. Higher values indicate stronger absorption, with typical values ranging from 0 to over 100,000 L·mol⁻¹·cm⁻¹ for highly absorbing chromophores [5].
Path Length (b) The path length represents the distance light travels through the sample solution, typically determined by the width of the cuvette used for measurement [1]. Standard cuvettes have a path length of 1 cm, though specialized cells with shorter path lengths (e.g., 1 mm) are available for highly concentrated samples to maintain absorbance within the ideal measurement range [2].
Concentration (c) The concentration of the absorbing species in the solution, usually expressed in moles per liter (mol·L⁻¹ or M). The Beer-Lambert Law assumes a linear relationship between concentration and absorbance, which holds true for dilute solutions but may deviate at higher concentrations due to molecular interactions [6].
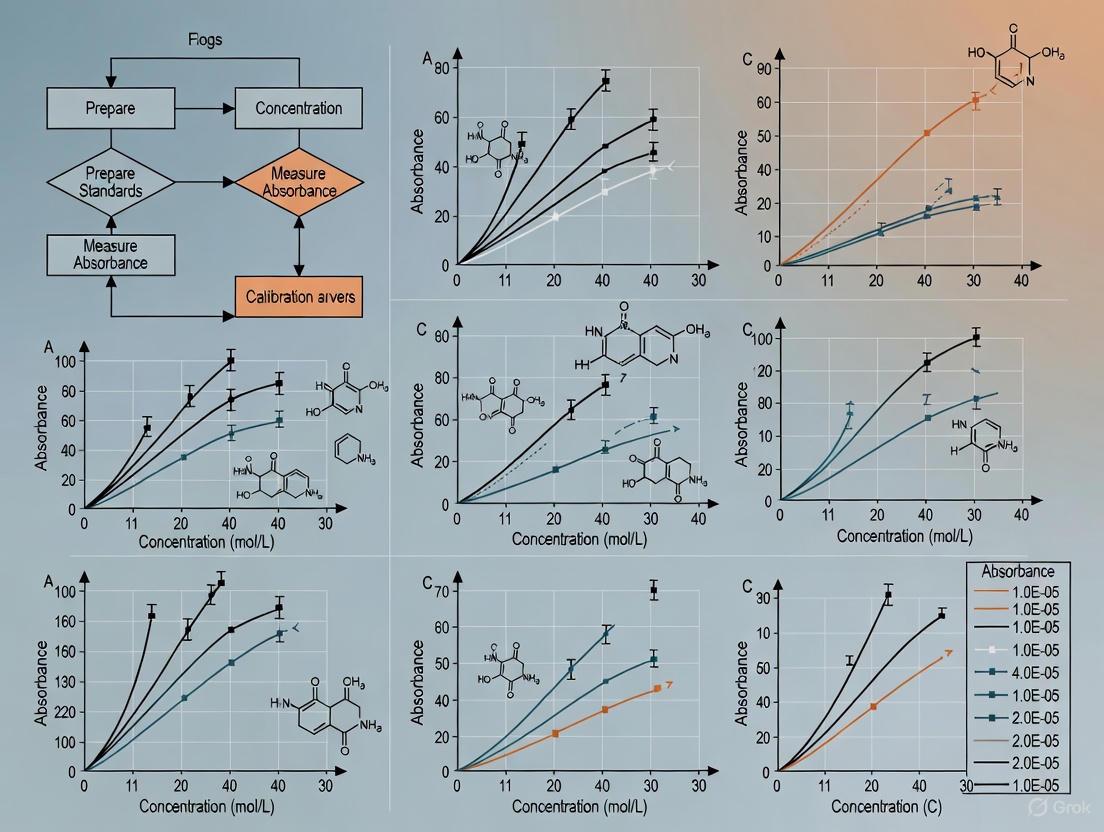
Figure 1: UV-Vis Spectrophotometer Workflow
Practical Application: Developing UV-Vis Calibration Curves
Protocol for Calibration Curve Development
Materials and Reagents
- Standard solutions of the analyte at known concentrations
- Appropriate solvent for preparing standard and sample solutions
- UV-transparent cuvettes (quartz for UV, glass or plastic for visible range)
- UV-Vis spectrophotometer with wavelength selection capability
- Volumetric flasks and pipettes for accurate solution preparation
Step-by-Step Procedure
Instrument Preparation
Standard Solution Preparation
- Prepare a stock solution of the analyte with accurately known concentration.
- Create a series of standard solutions covering the expected concentration range of your samples through serial dilution [4]. Ensure concentrations fall within the linear range of the Beer-Lambert relationship (typically absorbance values between 0.1 and 1.0 AU) [2].
Blank Measurement
- Fill a cuvette with the pure solvent used to prepare your standard and sample solutions.
- Place the cuvette in the sample holder and measure the blank to establish the I₀ reference value [2].
Standard Measurements
Calibration Curve Generation
- Plot absorbance (y-axis) versus concentration (x-axis) for all standard solutions.
- Perform linear regression analysis to obtain the equation of the best-fit line: y = mx + b, where m represents the slope (εb) and b is the y-intercept [4].
- Calculate the correlation coefficient (r²) to validate linearity; values ≥0.995 indicate acceptable linearity for quantitative analysis [4] [7].
Sample Analysis
- Measure the absorbance of unknown samples under identical conditions.
- Calculate sample concentrations using the regression equation from the calibration curve [1].
The Scientist's Toolkit: Essential Research Reagents and Materials
| Item | Function & Importance |
|---|---|
| Quartz Cuvettes | Optically transparent cells for holding samples; quartz is essential for UV measurements due to its transparency at short wavelengths [2]. |
| Standard Reference Materials | High-purity compounds of known concentration for preparing calibration standards and verifying method accuracy [4]. |
| Spectral Solvents | High-purity solvents with minimal UV absorption in the wavelength range of interest (e.g., water, acetonitrile, methanol) [2]. |
| Buffer Systems | Solutions for maintaining constant pH, particularly important for analytes whose absorption properties are pH-dependent [7]. |
| Chromogenic Reagents | Chemicals that react with target analytes to produce colored compounds with specific absorption maxima (e.g., promethazine for potassium bromate detection) [7] [8]. |
Table 2: Essential materials for UV-Vis spectrophotometric analysis
Method Validation Parameters
When developing UV-Vis methods for quantitative analysis, several validation parameters must be established to ensure reliability, accuracy, and precision [4] [7].
Linearity The calibration curve should demonstrate a directly proportional relationship between absorbance and concentration. The correlation coefficient (r²) provides a measure of linearity, with values ≥0.995 generally considered acceptable for quantitative analysis [4] [7].
Limit of Detection (LOD) and Limit of Quantification (LOQ)
- LOD: The lowest concentration of an analyte that can be detected, typically with a signal-to-noise ratio of 3:1 [4] [7]
- LOQ: The lowest concentration that can be quantified with acceptable precision and accuracy, typically with a signal-to-noise ratio of 10:1 [4] [7]
Accuracy and Precision
- Accuracy: Measured as percentage recovery, indicates how close the measured value is to the true value [4]. Recovery rates of 90-110% are generally acceptable [4] [7].
- Precision: The degree of agreement among repeated measurements, expressed as relative standard deviation (%RSD) [4]. Values <2% RSD typically indicate good precision [4].
The following validation data from recent research illustrates typical performance parameters:
| Validation Parameter | Result | Acceptance Criteria |
|---|---|---|
| Linear Range | 0.370-2.570 μg/mL | N/A |
| Regression Equation | Y = 0.020x + 0.030 | N/A |
| Correlation Coefficient (r²) | 0.9962 | ≥0.995 |
| Limit of Detection (LOD) | 0.005 μg/g | N/A |
| Limit of Quantification (LOQ) | 0.016 μg/g | N/A |
| Recovery Rate | 82.97-108.54% | 90-110% |
| Precision (%RSD) | 0.13% | <2% |
Table 3: Example method validation data for UV-Vis spectrophotometric determination [7]
Figure 2: UV-Vis Method Development and Validation Workflow
Applications in Pharmaceutical Research and Drug Development
The Beer-Lambert Law finds diverse applications in pharmaceutical research, quality control, and drug development processes:
API Quantification UV-Vis spectroscopy enables the quantification of active pharmaceutical ingredients (APIs) in raw materials, formulations, and dissolution media. For example, ascorbic acid content in beverage preparations was determined using a validated UV-Vis method with a standard vitamin C calibration curve, demonstrating 103.5% recovery with excellent precision (%RSD = 0.13%) [4].
Impurity Detection The technique can detect and quantify potentially harmful substances in pharmaceutical products. Recent research developed a green UV-Vis method for determining potassium bromate in bread using promethazine as a chromogenic reagent, achieving an LOD of 0.005 μg/g and LOQ of 0.016 μg/g [7] [8].
Dissolution Testing UV-Vis spectroscopy facilitates real-time monitoring of drug release from formulations during dissolution testing, providing critical data for biopharmaceutics classification and formulation optimization.
Biomolecule Analysis The method is widely employed for quantifying proteins, nucleic acids, and other biomolecules in drug discovery research, with specific applications in bacterial culturing, drug identification, and nucleic acid purity checks [2].
Limitations and Practical Considerations
Despite its widespread utility, the Beer-Lambert Law has limitations that researchers must consider for accurate quantitative analysis:
Deviations from Linearity The linear relationship between absorbance and concentration may deviate under certain conditions:
- High concentrations (>0.01 M): Molecular interactions and electrostatic effects can alter absorptivity [6]
- Chemical associations: Equilibrium processes such as dimerization or polymerization change effective molar absorptivity [6]
- Instrumental factors: Stray light, polychromatic radiation, and detector non-linearity can cause deviations [2] [6]
Scattering and Reflection Effects In real-world samples, light loss due to scattering (particularly in turbid solutions or biological tissues) and reflection at cuvette interfaces can lead to apparent deviations from the Beer-Lambert Law [9] [6]. For highly scattering media, modified versions of the law have been developed that incorporate differential pathlength factors to account for these effects [9].
Sample-Related Considerations
- Matrix effects: Complex sample matrices can cause apparent deviations through light scattering or interfering absorptions [8]
- pH dependence: Absorption spectra of ionizable compounds can shift significantly with pH changes [6]
- Temperature sensitivity: Molar absorptivity may vary with temperature, particularly for charge-transfer complexes [6]
Optimal Measurement Conditions To minimize errors and maintain linearity:
- Keep absorbance values between 0.1 and 1.0 AU [2]
- Use matched cuvettes with consistent path lengths [2]
- Employ high-purity solvents with minimal background absorption [2]
- Maintain constant temperature during measurements [6]
- Use monochromatic light with bandwidth narrower than the absorption band [6]
In the field of analytical chemistry, spectrophotometers are indispensable instruments for quantifying compound concentrations through UV-Vis spectroscopy. These instruments operate on the fundamental principle of measuring the absorption of ultraviolet or visible light by a sample, following the Beer-Lambert Law which states that absorbance is directly proportional to the concentration of the absorbing species [2]. The core components of a spectrophotometer include a stable light source, a wavelength selection system, a sample holder, and a detector [2]. The reliability of quantitative analysis, particularly in critical applications such as pharmaceutical drug development, hinges on selecting the appropriate spectrophotometer configuration and understanding its operational parameters. This application note provides a detailed comparison of single beam, double beam, and diode array spectrophotometers, with specific protocols for generating accurate UV-Vis calibration curves in compound quantification research.
Spectrophotometer Configurations: Principles and Comparison
Optical Designs and Operating Principles
Single Beam Spectrophotometers utilize the most straightforward optical design where a single light beam passes through the monochromator, through the sample, and to the detector [10]. This configuration requires separate measurements of the solvent blank (reference) and the sample, as the instrument cannot measure both simultaneously. The simplicity of this design makes it cost-effective but potentially susceptible to measurement drift from source instability.
Double Beam Spectrophotometers employ a mechanical chopper or beam splitter to divide the light from the source into two separate paths: a reference beam and a sample beam [11]. This design allows simultaneous measurement of the sample and reference, with the detector alternating between the two beams [11] [10]. The key advantage lies in the instrument's ability to automatically compensate for solvent absorption and source intensity fluctuations in real-time, providing enhanced stability and reliability [11].
Diode Array Spectrophotometers represent a significant advancement in detection technology. Instead of using a monochromator before the sample, these instruments pass polychromatic light through the sample and then disperse it onto an array of photodiodes [12]. This enables simultaneous detection of all wavelengths across the spectrum, dramatically reducing acquisition time and allowing for full spectral capture of chromatographic peaks [12]. The reversed optical path distinguishes this configuration from scanning monochromator-based systems.
Table 1: Comparative Analysis of Spectrophotometer Configurations
| Parameter | Single Beam | Double Beam | Diode Array |
|---|---|---|---|
| Optical Design | Single light path | Split beam: reference & sample | Polychromatic light with post-dispersion |
| Measurement Sequence | Sequential blank & sample | Simultaneous reference & sample | Simultaneous all wavelengths |
| Data Acquisition Speed | Moderate | Fast | Very fast (full spectrum in seconds) |
| Stability & Compensation | Susceptible to source drift | Real-time compensation for drift [11] | Stable, but different compensation approach |
| Wavelength Selection | Pre-sample monochromator | Pre-sample monochromator | Post-sample polychromator |
| Spectral Resolution | Dependent on monochromator slit width | Dependent on monochromator slit width | Determined by diode density and optics |
| Primary Applications | Routine quantitative analysis at fixed wavelengths | Kinetic studies, wavelength scanning [11] | Spectral scanning, peak purity assessment [12] |
| Approximate Cost | Low | Medium to High | High |
Performance Characteristics and Selection Criteria
The choice between spectrophotometer configurations depends heavily on the specific requirements of the quantification method. Double beam instruments offer superior stability because their readings are not easily affected by external factors such as energy and voltage fluctuations, lamp drift, and stray light [11]. This makes them particularly suitable for applications requiring high precision and for experiments extending over prolonged periods. Additionally, double beam spectrophotometers require minimal warmup time, which increases throughput and prolongs the lamp's lifespan [11].
Diode array detectors provide significant advantages for method development and peak purity assessment because they capture the entire UV spectrum simultaneously [12]. This capability is invaluable for identifying compounds based on their spectral characteristics and for detecting potential impurities in analytical samples. The ability to retrospectively analyze data at different wavelengths without reinjection saves considerable time in method development.
Table 2: Advantages and Limitations of Different Spectrophotometer Configurations
| Configuration | Key Advantages | Key Limitations |
|---|---|---|
| Single Beam | Simple operation, lower cost, compact size | Requires separate blank measurement, susceptible to source drift, slower throughput |
| Double Beam | High stability, real-time blank correction, fast scanning [11] | Higher cost, more complex operation [11] |
| Diode Array | Rapid full spectrum acquisition, peak purity assessment [12] | Higher cost, potentially lower resolution depending on design |
Experimental Protocols for UV-Vis Calibration Curve Generation
General Preparation and Instrument Setup
Research Reagent Solutions and Essential Materials:
Table 3: Essential Research Reagents and Materials
| Item | Function/Specification |
|---|---|
| High-Purity Analytical Standard | Primary reference material of the target compound with known purity |
| Appropriate Solvent | HPLC-grade solvent transparent in the spectral region of interest |
| Volumetric Flasks | Class A, various sizes for standard solution preparation |
| Cuvettes | Quartz for UV range (190-380 nm); glass or plastic for visible range [2] |
| Buffer Salts | For maintaining stable pH when required by analyte properties |
Protocol 1: Instrument Startup and Qualification
- Power on the spectrophotometer and allow the lamp to warm up for the manufacturer-recommended time (typically 15-30 minutes; double beam instruments may require less warmup time [11]).
- Select the appropriate measurement mode (absorbance) and set the analytical wavelength based on the compound's λmax.
- For diode array systems, set the spectral acquisition range (typically 200-800 nm for full UV-Vis coverage).
- Perform instrument qualification using certified holmium oxide or didymium filters to verify wavelength accuracy.
- Using a matched pair of cuvettes, fill both with the blank solvent and perform a baseline correction to account for any solvent absorption or cuvette differences.
Standard Solution Preparation and Measurement
Protocol 2: Stock and Working Standard Preparation
- Accurately weigh an appropriate amount of the analytical standard using an analytical balance.
- Transfer quantitatively to a volumetric flask and dilute to volume with the selected solvent to create the stock standard solution.
- Calculate the stock solution concentration using the formula: C_stock = (mass × purity) / volume.
- Create a series of working standards by performing serial dilutions of the stock solution to cover the expected concentration range. Typically, 5-8 concentration levels are recommended for a calibration curve.
- Ensure all solutions are properly mixed and free of air bubbles before measurement.
Protocol 3: Absorbance Measurement and Data Collection
- Set the instrument to zero absorbance using the blank solvent contained in a cuvette identical to those used for samples.
- For single beam instruments: Measure each standard solution in sequence, rinsing the cuvette at least three times with the next standard to be measured.
- For double beam instruments: Place the blank in the reference position and measure each standard in the sample position [11].
- For diode array instruments: Acquire full spectra for each standard, noting the absorbance at the analytical wavelength.
- Record all absorbance values, ensuring they fall within the instrument's linear range (typically 0.2-1.0 AU for optimal performance).
Data Analysis and Quality Control
Protocol 4: Calibration Curve Generation and Validation
- Plot absorbance (y-axis) versus concentration (x-axis) for all standard solutions.
- Perform linear regression analysis to obtain the equation: y = mx + b, where m is the slope and b is the y-intercept.
- Calculate the correlation coefficient (R²) to assess linearity; for quantitative work, R² should typically be ≥0.995.
- Determine the linear dynamic range by identifying where the response deviates from linearity (typically at higher concentrations).
- Calculate the limit of detection (LOD = 3.3σ/S) and limit of quantification (LOQ = 10σ/S), where σ is the standard deviation of the blank and S is the slope of the calibration curve.
Protocol 5: Sample Analysis and Quantification
- Prepare unknown samples in the same solvent system as the standards.
- Measure the absorbance of unknown samples using the same instrumental conditions as the calibration standards.
- Calculate the unknown concentration using the regression equation: Cunknown = (Aunknown - b) / m.
- For samples falling outside the calibration range, appropriately dilute and remeasure, applying the dilution factor in final concentration calculations.
- Include quality control samples with known concentrations to verify method accuracy throughout the analysis.
Advanced Applications in Pharmaceutical Research
Specialized Quantification Methods
In drug development, each spectrophotometer configuration offers unique advantages for specific applications. Double beam systems excel in kinetic studies where reaction progress is monitored over time at a specific wavelength, as their inherent stability minimizes baseline drift during extended measurements [11]. Diode array systems are particularly valuable for method development because they enable retrospective analysis at different wavelengths without reinjection and facilitate peak purity assessment by comparing spectra across a chromatographic peak [12].
For DNA and protein quantification, double beam spectrophotometers provide the rapid, reproducible measurements essential for high-throughput applications [11]. The simultaneous reference measurement capability allows for accurate ratio-based calculations (e.g., A260/A280 for nucleic acid purity) without concern for source fluctuation between measurements.
Troubleshooting and Method Validation
Common Issues and Solutions:
- Poor Linear Range: Ensure absorbance readings remain between 0.2-1.0 AU; dilute samples as needed.
- Baseline Drift: More common in single beam systems; allow sufficient lamp warmup time and maintain constant temperature.
- Spectral Noise: More pronounced at narrower bandwidths; use appropriate slit widths balancing resolution and signal-to-noise ratio.
Method Validation Parameters: For regulatory applications such as pharmaceutical quality control, method validation should include assessment of linearity, accuracy, precision, LOD, LOQ, and robustness. The higher precision achievable with UV detection (<0.2% RSD) is particularly important in pharmaceutical testing where typical potency specifications for drug substances range from 98.0% to 102.0% [12].
The selection of an appropriate spectrophotometer configuration is critical for developing robust UV-Vis calibration methods in compound quantification research. Single beam instruments offer cost-effectiveness for routine fixed-wavelength analyses, while double beam configurations provide enhanced stability for dynamic experiments and scanning applications. Diode array systems deliver unparalleled speed and spectral information for method development and peak purity assessment. By following the detailed protocols outlined in this application note, researchers can generate reliable calibration curves that meet the rigorous demands of pharmaceutical development and other quantitative analytical applications. The implementation of proper quality control measures and understanding of each instrument's capabilities and limitations will ensure accurate, reproducible results in compound quantification studies.
The Critical Role of Calibration Curves in Quantitative Analysis
In the realm of quantitative analytical science, the calibration curve, also known as a standard curve, serves as a fundamental cornerstone for determining the concentration of unknown substances. This methodological approach establishes a predictable relationship between the instrumental response and the analyte concentration, allowing researchers to convert measurable signals into meaningful quantitative data [13]. In the specific context of UV-Vis spectrophotometry, this technique leverages the principle that the absorbance of light by a chemical species is directly proportional to its concentration, as described by the Beer-Lambert law [13] [14].
The critical importance of calibration curves extends across numerous scientific disciplines, including pharmaceutical quality control, environmental monitoring, and biomedical research [13] [14]. For researchers and drug development professionals, proper calibration ensures the accuracy, precision, and reliability of quantitative measurements, which form the basis for critical decisions regarding compound characterization, dosage formulation, and regulatory compliance [15]. Without robust calibration methodologies, the validity of experimental results remains questionable, potentially compromising research outcomes and product safety.
Theoretical Foundation
Fundamental Principles of UV-Vis Spectrophotometry
Ultraviolet-Visible (UV-Vis) spectrophotometry operates on the principle that molecules absorb light in the ultraviolet and visible regions of the electromagnetic spectrum. When a sample is exposed to UV-Vis light, chromophores within the molecules undergo electronic transitions, absorbing specific wavelengths of light [13]. A UV-Vis spectrophotometer consists of several key components: a light source (typically a xenon lamp, or combination of tungsten/halogen and deuterium lamps), a wavelength selector (monochromator or filters), a sample holder (cuvette), and a detector [14].
The instrument measures the transmittance (the percentage of light passing through the sample) and calculates the absorbance according to the mathematical relationship A = -log(T), where T is transmittance [14]. According to the Beer-Lambert law, absorbance (A) is directly proportional to the concentration (c) of the absorbing species: A = εMc, where ε is the molar absorptivity or extinction coefficient, M is the path length of the cuvette, and c is the concentration [13]. This linear relationship between absorbance and concentration forms the theoretical basis for quantitative analysis using calibration curves.
Mathematical Modeling of Calibration Curves
The fundamental mathematical model for a calibration curve in UV-Vis spectrophotometry is a linear relationship expressed as:
S = kC + b
Where S is the measured signal (absorbance), C is the analyte concentration, k is the sensitivity (slope), and b is the y-intercept [16]. This model assumes a first-order dependence of the signal on concentration. The sensitivity (k) represents the change in signal per unit change in concentration, while the intercept (b) ideally should be close to zero, though instrumental background or matrix effects may cause slight deviations [16].
The validity of this linear model must be experimentally verified across the concentration range of interest, as deviations from linearity may occur at higher concentrations due to instrumental limitations or chemical factors such as molecular associations [16]. For quantitative analysis, the coefficient of determination (R²) is used to evaluate the goodness of fit of the experimental data to the linear regression model, with values closer to 1.0 indicating a better fit [14].
Figure 1: Theoretical foundation of calibration curves showing the relationship between fundamental principles and practical application.
Performance Parameters for Method Validation
Critical Validation Parameters
For any quantitative analytical method, rigorous validation is essential to ensure the reliability and accuracy of results. Instrument validation for UV-Vis spectrophotometers encompasses multiple performance parameters that collectively determine the suitability of the method for quantitative analysis [17]. These parameters, as prescribed in standards such as JIS K0115 "General rules for molecular absorptiometric analysis," provide a comprehensive framework for assessing instrument performance [17].
Wavelength accuracy refers to the agreement between the instrument's measured wavelength values and the true wavelength values, typically verified using emission lines of deuterium or low-pressure mercury lamps or absorption peaks of certified reference materials [17]. Photometric accuracy assesses the correctness of absorbance or transmittance measurements, while photometric repeatability evaluates the precision of replicate measurements [17]. Stray light, defined as light outside the specified wavelength that reaches the detector, can significantly impact measurement accuracy, particularly at high absorbance values [17].
Comprehensive Performance Specifications
Table 1: Key performance parameters for UV-Vis spectrophotometer validation
| Performance Parameter | Definition | Impact on Quantitative Analysis | Acceptance Criteria |
|---|---|---|---|
| Wavelength Accuracy | Agreement between measured and true wavelength values | Affects spectral identification and selectivity | Typically ±0.1 nm for high-performance instruments [17] |
| Photometric Accuracy | Correctness of absorbance/transmittance measurements | Directly impacts concentration accuracy | Dependent on application requirements [17] |
| Stray Light | Light outside specified wavelength reaching detector | Causes non-linearity at high absorbance | Critical for high-absorbance samples [17] |
| Noise Level | Random fluctuations in measured signal | Affects detection and quantitation limits | Lower noise enables better detection of small peaks [17] |
| Baseline Flatness | Deviation from flat baseline across wavelength range | Impacts measurement consistency | Should be minimal across analytical range [17] |
The noise level, defined as the maximum deviation of absorbance measured over time, serves as an important indicator of instrument condition, particularly the state of the light source [17]. As lamps deteriorate over time, noise typically increases, adversely affecting measurement precision [17]. Baseline stability and flatness further contribute to measurement quality by ensuring consistent performance across the analytical wavelength range [17].
Experimental Protocols
Materials and Equipment
Table 2: Essential research reagents and materials for calibration curve preparation
| Item | Specification | Function/Purpose |
|---|---|---|
| Standard Solution | High-purity analyte in appropriate solvent | Provides known concentrations for calibration [14] |
| Solvent | Deionized water or HPLC-grade organic solvents | Matrix for preparing standards and samples [14] |
| Volumetric Flasks | Class A, various volumes (e.g., 10 mL, 25 mL, 50 mL) | Precise preparation of standard solutions [14] |
| Pipettes and Tips | Calibrated, appropriate volume range | Accurate transfer of solutions [14] |
| Cuvettes | Quartz (UV) or glass (Vis), matched pathlength | Sample holder for spectrophotometer [14] |
| UV-Vis Spectrophotometer | Validated performance parameters | Instrument for absorbance measurements [17] [14] |
Step-by-Step Calibration Curve Protocol
Preparation of Standard Solutions
Begin by preparing a concentrated stock solution of the standard compound using an analytical balance and volumetric flask to ensure precise concentration [14]. The solvent should be identical to that used for unknown samples to maintain matrix matching. Prepare a series of standard solutions spanning the expected concentration range of unknown samples through serial dilution [14]. A minimum of five standard concentrations is recommended to establish a reliable calibration curve, with concentrations appropriately spaced to define the concentration-response relationship [14].
For the serial dilution, pipette a specific volume of the stock solution into the first volumetric flask and dilute to volume with solvent. Mix thoroughly, then transfer an aliquot from this solution to the next flask and repeat the process. This systematic approach ensures accurate preparation of decreasing standard concentrations while maintaining consistent matrix composition [14].
Absorbance Measurement and Data Collection
Transfer each standard solution to an appropriate cuvette, ensuring compatibility with the spectrophotometer's wavelength range (quartz for UV measurements, glass or plastic for visible range) [14]. Measure the absorbance of each standard solution at the predetermined analytical wavelength, using solvent as the blank to zero the instrument [14]. Obtain multiple readings (typically 3-5 replicates) for each standard to assess measurement precision and enable statistical evaluation of the data [14].
Repeat the measurement process for unknown samples prepared in the same matrix as the standards. Maintain consistent measurement conditions (temperature, timing, instrument parameters) throughout the analysis to minimize variability. Record all absorbance values systematically, noting any deviations from expected values or observations during measurement.
Figure 2: Experimental workflow for developing and applying calibration curves in quantitative analysis.
Data Analysis and Interpretation
Construction of the Calibration Curve
Following data collection, plot the average absorbance values for each standard on the y-axis against the corresponding known concentrations on the x-axis [14]. The resulting graph should display a linear relationship across the working concentration range, with deviations from linearity potentially occurring at higher concentrations (limit of linearity) due to detector saturation or deviations from the Beer-Lambert law [14].
Apply linear regression analysis to the data points using appropriate statistical software, generating the equation y = mx + b, where y represents absorbance, m is the slope (sensitivity), x is the concentration, and b is the y-intercept [14]. The slope (m) of the calibration curve reflects the sensitivity of the method, with steeper slopes indicating greater sensitivity to concentration changes. The y-intercept (b) should theoretically pass through the origin (zero absorbance at zero concentration), though minor deviations may occur due to matrix effects or instrumental background [14].
Assessment of Curve Quality and Validation
Evaluate the quality of the calibration curve using the coefficient of determination (R²), which quantifies the goodness of fit of the experimental data to the linear regression model [14]. While R² values close to 1.0 (typically >0.995 for quantitative work) indicate a good fit, this parameter alone does not guarantee analytical suitability [15]. Additional statistical measures, including residual analysis and examination of homoscedasticity, provide deeper insight into the appropriateness of the linear model [15].
The phenomenon of heteroscedasticity, where the variance of measurements changes with concentration, is common in analytical data and should be addressed through appropriate weighting factors in the regression model if significant [15]. For proper method validation, include quality control (QC) samples with known concentrations across the calibration range to verify the accuracy and precision of the established curve [15]. The calibration curve should be reconstructed with each analytical batch to account for potential instrument drift over time [15].
Advanced Considerations and Troubleshooting
Matrix Effects and Mitigation Strategies
In quantitative analysis, matrix effects represent a significant challenge, occurring when components in the sample matrix enhance or suppress the analytical signal, leading to inaccurate concentration determinations [15]. These effects are particularly problematic in complex biological matrices where co-eluting compounds may interfere with the target analyte [15].
To mitigate matrix effects, several strategies can be employed. The use of matrix-matched calibrators, where standards are prepared in a matrix similar to the unknown samples, helps minimize differences between calibration and sample matrices [15]. For endogenous analytes, creating a "blank" matrix through stripping techniques (e.g., charcoal treatment, dialysis) or using synthetic matrices can provide appropriate calibration media, though commutability between the calibrator matrix and native patient samples must be verified [15]. The incorporation of stable isotope-labeled internal standards (SIL-IS) for each target analyte represents the most effective approach for compensating for matrix effects, as these compounds experience nearly identical ionization suppression/enhancement as the native analytes while being distinguishable mass spectrometrically [15].
Troubleshooting Common Issues
Several common issues may arise during calibration curve development that require troubleshooting. Non-linearity at higher concentrations may indicate detector saturation, stray light effects, or chemical associations such as dimerization [17] [14]. This can be addressed by diluting samples, using a shorter pathlength cuvette, or restricting the analytical range. Poor reproducibility between replicates often results from instrumental issues (e.g., lamp degradation, excessive noise) or solution handling problems (incomplete mixing, pipetting errors) [17].
Abnormal intercept values significantly different from zero may suggest contamination in reagents, incorrect blank preparation, or non-specific interference in the matrix [16]. Inconsistent QC sample recovery might indicate calibration curve instability, matrix effects differences between standards and samples, or analyte degradation [15]. Regular instrument validation, including checks of wavelength accuracy, photometric accuracy, and stray light, helps identify and correct instrumental contributions to these issues [17].
Application in Drug Development and Research
In pharmaceutical research and development, calibration curves play an indispensable role in multiple stages of the drug development pipeline. During drug discovery, they facilitate the quantification of lead compounds in biological matrices for preliminary pharmacokinetic assessments. In preclinical development, validated calibration methods enable accurate determination of drug concentrations in plasma, tissues, and excreta for comprehensive ADME (Absorption, Distribution, Metabolism, and Excretion) studies [15].
For bioavailability and bioequivalence studies, rigorously validated calibration curves with appropriate matrix matching are essential for generating reliable pharmacokinetic data [15]. In formulation development, UV-Vis spectrophotometry with proper calibration supports the assessment of drug stability, solubility, and release profiles from dosage forms. The implementation of Good Laboratory Practice (GLP) and Good Manufacturing Practice (GMP) guidelines requires documented calibration procedures and regular performance verification to ensure data integrity and regulatory compliance [17] [15].
The critical role of calibration curves extends to quality control laboratories, where they are employed for assay determination, impurity quantification, and content uniformity testing of pharmaceutical products [13]. In these regulated environments, calibration methods must be thoroughly validated according to regulatory guidelines, with defined acceptance criteria for accuracy, precision, linearity, and range [15]. The selection of appropriate calibration standards, matrix considerations, and statistical evaluation of curve fit all contribute to the overall quality and reliability of analytical results supporting drug development and manufacturing.
Ultraviolet-visible (UV-Vis) spectroscopy is a fundamental analytical technique used to determine the concentration of compounds in solution [18]. The successful quantification of analytes, particularly in critical fields like drug development, relies on a thorough understanding of three core parameters: molar absorptivity, path length, and dynamic range. These parameters are intrinsically linked through the Beer-Lambert law, which forms the theoretical basis for absorption spectroscopy [19] [20]. This application note details the definition, relationship, and practical application of these parameters within the context of developing robust UV-Vis calibration curves for compound quantification research, providing scientists with the protocols needed to generate reliable and reproducible data.
Theoretical Foundations
The Beer-Lambert Law
The relationship between light absorption and the properties of a solution is quantitatively described by the Beer-Lambert Law. The equation is expressed as:
A = εbc
Where:
- A is the measured Absorbance (unitless)
- ε is the Molar Absorptivity (L·mol⁻¹·cm⁻¹)
- b is the Path Length (cm)
- c is the Concentration (mol·L⁻¹) [19] [18] [20]
This law states that the absorbance of light by a solution is directly proportional to the concentration of the absorbing species and the path length the light travels through the sample [19].
In-Depth Parameter Analysis
Molar Absorptivity (ε)
Molar absorptivity, also known as the extinction coefficient, is a physical constant that defines how strongly a chemical species absorbs light at a specific wavelength [20] [21]. It is a measure of the amount of light absorbed per unit concentration [20]. Its value is influenced by the chemical identity of the analyte and the solvent, as well as environmental factors such as temperature and pH [21].
- Significance: A compound with a high molar absorptivity is very effective at absorbing light, allowing for its detection at lower concentrations [20] [21]. Values can vary significantly between compounds and are typically on the order of 10⁵ L·mol⁻¹·cm⁻¹ for simple molecules [19].
- Dependence: Because its value can be affected by the chemical environment (e.g., solvent, pH, temperature) and instrument-specific factors, it is often determined empirically through calibration curves rather than relied upon from literature [19] [21].
Path Length (b)
The path length is the distance that light travels through the sample solution, typically measured in centimeters (cm) [19]. In standard spectroscopy, this is determined by the width of the cuvette, with 1 cm being the most common [18].
- Significance: Absorbance increases in direct proportion to the path length [19] [22]. This property can be leveraged analytically; for very dilute samples, using a long pathlength cell (e.g., 50 mm or 100 mm) can increase the absorbance signal, making quantification more reliable [23] [22]. Conversely, for very concentrated samples, a shorter path length can bring an otherwise off-scale absorbance into the measurable range.
- Variable Pathlength Technique: This method involves measuring absorbance at multiple path lengths for a single concentration. A plot of Absorbance vs. Path Length yields a straight line whose slope (m) is equal to the product of the molar absorptivity and the concentration (m = εc). This technique averages out minor inconsistencies and can negate the need for background subtraction [23].
Dynamic Range
The dynamic range in UV-Vis spectroscopy refers to the concentration interval over which a change in concentration produces a proportional (linear) change in the measured absorbance, in accordance with the Beer-Lambert Law [14].
- Limits: The lower limit of the dynamic range is determined by the limit of detection (LOD), the smallest concentration that can be reliably distinguished from background noise. The upper limit is the limit of linearity (LOL), the concentration at which the instrument begins to saturate and the absorbance-concentration relationship deviates from linearity [14].
- Practical Impact: A calibration curve is considered valid only within the dynamic range of the instrument for that specific analyte under the specified conditions [14]. Attempting to quantify a sample outside this range will lead to significant errors.
Table 1: Key Parameters in UV-Vis Quantification
| Parameter | Symbol & Units | Definition | Role in Beer's Law | Practical Consideration |
|---|---|---|---|---|
| Molar Absorptivity | ε (L·mol⁻¹·cm⁻¹) | Measure of how strongly a species absorbs light at a given wavelength [20] [21]. | Proportionality constant linking absorption to concentration [19]. | Compound-specific; can be affected by solvent, pH, and temperature [21]. |
| Path Length | b (cm) | Distance light travels through the sample solution [19]. | Directly proportional to absorbance [19]. | Can be varied using specialized cells to adjust absorbance for low/high concentrations [23] [22]. |
| Dynamic Range | - | Concentration range over which absorbance response is linear [14]. | Defines the valid range for the equation A = εbc. | Calibration curves are only valid within this range; limits are LOD and LOL [14]. |
Experimental Protocols
Protocol 1: Establishing a UV-Vis Calibration Curve
This protocol provides a step-by-step methodology for creating a calibration curve to quantify an unknown sample, a cornerstone technique in analytical chemistry and biochemistry [14] [13].
Research Reagent Solutions & Essential Materials
- Personal Protective Equipment (PPE): Gloves, lab coat, and safety glasses [14].
- Standard Solution: A concentrated stock solution of the pure analyte with a known, accurately determined concentration [14].
- Solvent: High-purity solvent (e.g., deionized water, methanol) matching the solvent of the unknown sample [14].
- Pipettes and Tips: Calibrated precision pipettes and corresponding tips for accurate liquid handling [14].
- Volumetric Flasks/Microtubes: For preparing standard solutions with precise volumes [14].
- UV-Vis Spectrophotometer: Instrument with a light source, wavelength selector, and detector [18] [14].
- Cuvettes: Sample holders compatible with the spectrophotometer and the wavelength range (e.g., quartz for UV) [14].
- Computer: For instrument control, data collection, and analysis [14].
Procedure:
- Prepare Stock Solution: Accurately weigh the solute and transfer it to a volumetric flask. Dilute to the mark with solvent to create a concentrated stock solution [14].
- Generate Standard Solutions: Perform a serial dilution to create a minimum of five standard solutions spanning the expected concentration range of the unknown. For example, prepare standards at 100%, 75%, 50%, 25%, and 10% of the stock concentration. Use volumetric flasks and change pipette tips between each transfer to ensure accuracy [14].
- Prepare Samples: Transfer the standard solutions and the unknown sample(s) into clean, labeled cuvettes. Ensure the unknown samples are prepared using the same buffer and pH as the standards [14].
- Measure Absorbance:
- Zero (blank) the spectrophotometer using a cuvette filled only with solvent [18].
- Place each standard solution in the spectrophotometer and measure the absorbance at the predetermined analytical wavelength (typically at an absorbance maximum).
- Obtain between three and five replicate readings for each standard to assess precision [14].
- Repeat the measurement process for the unknown sample(s).
- Plot Data and Analyze: Plot the average absorbance (y-axis) against the known concentration (x-axis) for each standard. Use statistical software to fit the data to a linear regression (y = mx + b), and determine the coefficient of determination (R²) to assess the goodness of fit. An R² value of 0.9 or better is typically acceptable [18] [14].
The following workflow diagram illustrates the key steps in this protocol:
Protocol 2: Leveraging Variable Path Length for Slope Spectroscopy
This method is particularly useful for obtaining accurate concentration data without the need for a full calibration curve or when sample consistency is variable [23].
Research Reagent Solutions & Essential Materials
- All materials from Protocol 1, except:
- Variable Pathlength Cell: A sample holder whose path length can be accurately and reproducibly varied [23].
Procedure:
- Prepare Sample: Place the sample of unknown concentration into the variable pathlength cell [23].
- Measure at Multiple Path Lengths: Without changing the sample concentration, measure the absorbance at several different, accurately known path lengths [23].
- Plot and Calculate Slope: Plot the measured absorbance (y-axis) against the path length (x-axis). This should yield a straight line. Perform a linear regression to determine the slope (m) of the line [23].
- Determine Concentration: The slope (m) from the plot is equal to the product of the molar absorptivity and the concentration (m = εc). If the molar absorptivity (ε) is known for the compound, the concentration (c) can be calculated directly as c = m / ε [23].
Data Analysis and Application
Table 2: Quantitative Relationships and Ranges for Key Parameters
| Parameter | Typical Units | Proportionality | Typical Values / Range | Analytical Impact |
|---|---|---|---|---|
| Molar Absorptivity (ε) | L·mol⁻¹·cm⁻¹ | Directly proportional to A [19]. | Up to ~100,000 for simple molecules [19]. | High ε enables lower detection limits [20] [21]. |
| Path Length (b) | cm | Directly proportional to A [19]. | Standard: 1 cm; Long-path: 2, 5, 10 cm [22]. | 10x longer path → 10x higher A for same concentration [22]. |
| Coefficient of Determination (R²) | Unitless | Measures linearity of calibration curve [14]. | >0.9 for an acceptable calibration [18]. | Quantifies reliability of the calibration model [14]. |
Visualizing Parameter Relationships
The following diagram illustrates the core relationship between the parameters of the Beer-Lambert Law and the experimental techniques used to manipulate them for accurate quantification.
A rigorous understanding of molar absorptivity, path length, and dynamic range is non-negotiable for developing precise and accurate UV-Vis calibration methods in quantitative research. Molar absorptivity provides the fundamental link between a compound's chemical structure and its light-absorbing properties. Path length offers a practical tool to optimize analytical signals for a wide range of concentrations. Finally, the dynamic range defines the operational limits within which the Beer-Lambert Law holds true. By applying the detailed protocols and principles outlined in this application note, researchers and drug development professionals can ensure their spectroscopic data is robust, reliable, and suitable for critical decision-making processes.
Ultraviolet-visible (UV-Vis) spectroscopy is a fundamental analytical technique used for the quantitative analysis of compounds in solution. The reliability of this quantification is critically dependent on the quality of the calibration curve, which itself is heavily influenced by the proper selection and handling of samples and solvents. This document outlines key considerations and detailed protocols for ensuring optimal absorbance measurements within the context of developing robust UV-Vis calibration curves for compound quantification research, particularly relevant to drug development.
The foundational principle of UV-Vis quantification, the Beer-Lambert Law (A = εbc), establishes a linear relationship between absorbance (A) and the concentration (c) of an analyte in solution [18]. However, this relationship can be compromised by inappropriate sample and solvent choices, leading to inaccurate concentration predictions. This note provides a structured approach to mitigate these risks.
Fundamental Principles and the Impact of Sample & Solvent
The accuracy of a calibration curve begins with understanding how the measurement environment—the solvent and the sample itself—affects light absorption.
The Beer-Lambert Law and the Solvent Blank
The Beer-Lambert Law is the cornerstone of absorbance quantification [18]. A critical, often overlooked, step in its application is the use of a blank reference to zero the instrument. The blank must contain the same solvent and any other chemical components present in the sample, except for the analyte of interest. This corrects for any light absorption or scattering caused by the solvent or cuvette, ensuring that the measured absorbance is due solely to the target compound [18].
Sample Limitations
UV-Vis spectroscopy performs best with true solutions. As noted in the search results, if a sample is more of a suspension of solid particles in a liquid, the sample will scatter light more than absorb it, leading to skewed and unreliable data [18]. While accessories for solid samples exist, the technique is most efficient and accurate for liquids and solutions.
Critical Considerations for Solvents and Samples
The following table summarizes the key factors researchers must evaluate when selecting and preparing solvents and samples for UV-Vis calibration.
Table 1: Critical Solvent and Sample Considerations for UV-Vis Absorbance Measurements
| Factor | Consideration | Impact on Absorbance Measurement |
|---|---|---|
| Solvent Transparency | The solvent must not absorb significantly in the spectral region where the analyte absorbs. | A solvent with a high background absorbance will reduce the available light path, decreasing the signal-to-noise ratio and the dynamic range for detection [14]. |
| Solvent-Analyte Chemical Compatibility | The solvent must fully dissolve the analyte without reacting with it. | Incomplete dissolution can cause light scattering. Chemical reactions can alter the analyte's chemical structure and its absorptivity (ε), invalidating the calibration [14]. |
| Matrix Effects | For complex samples (e.g., biological fluids), other components in the sample can scatter light or absorb at the same wavelength as the analyte. | This can cause signal suppression or enhancement, leading to a non-linear calibration curve and inaccurate quantification of the unknown [15]. |
| Sample Homogeneity | The sample must be a clear, homogeneous solution. | Suspended particles or turbidity cause significant light scattering, which is measured as absorbance, leading to a positive bias in the concentration calculation [18]. |
| Pathlength | The distance light travels through the sample (typically defined by the cuvette). | According to the Beer-Lambert Law, absorbance is directly proportional to pathlength. Using a consistent, appropriate pathlength is crucial for accurate calibration [18]. |
Advanced Consideration: Mitigating Matrix Effects
In bioanalysis, where samples are complex matrices like plasma, a "blank matrix" is used to prepare calibration standards. This is a material (e.g., drug-free plasma) that is as representative as possible of the sample matrix but devoid of the analyte. Using matrix-matched calibrators helps to compensate for matrix effects, ensuring the signal-to-concentration relationship is conserved between standards and unknown samples [15]. For endogenous analytes where a true blank is unavailable, the method of standard additions can be applied [24].
Experimental Protocol: Developing a UV-Vis Calibration Curve
This protocol provides a detailed methodology for preparing standards and generating a reliable calibration curve for compound quantification.
The Scientist's Toolkit: Essential Materials
Table 2: Key Research Reagent Solutions and Essential Materials
| Item | Function/Explanation |
|---|---|
| Personal Protective Equipment (PPE) | Protects the researcher from exposure to hazardous chemicals and samples [14]. |
| High-Purity Analytical Standard | A solution with a known, high concentration of the pure analyte. Serves as the source for preparing all calibration standards [14]. |
| UV-Transparent Solvent | A solvent (e.g., HPLC-grade water, methanol, acetonitrile) that does not absorb in the UV-Vis range of interest, ensuring analyte signal is not obscured [14]. |
| Precision Pipettes and Calibrated Tips | Allows for accurate and precise measurement and transfer of liquid volumes, which is critical for preparing standards of exact concentrations [14]. |
| Volumetric Flasks | Used for precise preparation of standard solutions to ensure accuracy in concentration [14]. |
| UV-Vis Spectrophotometer | The instrument used to measure the absorbance of light by the standard and unknown samples [18] [14]. |
| Spectrophotometer Cuvettes | Sample holders that must be clean and matched. Quartz is required for UV range measurements due to its transparency at short wavelengths [14]. |
Step-by-Step Workflow
The following diagram illustrates the overall workflow for creating and validating a UV-Vis calibration curve.
Step 1: Prepare a Concentrated Stock Solution
- Accurately weigh the solute and transfer it to a volumetric flask.
- Dilute to the mark with the appropriate solvent to create a stock solution of known, high concentration [14].
Step 2: Prepare Calibration Standards via Serial Dilution
- A minimum of five standards are recommended for a good calibration curve [14].
- Label a series of volumetric flasks or microtubes with the target concentrations. The standards should span the expected concentration range of the unknown samples, from just above the estimated unknown concentration down to an order of magnitude lower [18].
- Perform a serial dilution:
- Pipette a specific volume of the stock solution into the first flask.
- Dilute to the mark with solvent and mix thoroughly.
- Pipette from this first dilution into the next flask.
- Repeat the process of dilution and transfer to create the series of standards [14].
Step 3: Measure Absorbance of Standards and Unknowns
- Zero the Instrument: Place a cuvette filled only with the solvent (the blank) into the spectrophotometer and set the absorbance to zero. This critical step must be performed before reading any standards or unknowns [18].
- Measure Standards: Transfer each standard to a clean cuvette and obtain an absorbance reading at the predetermined wavelength of maximum absorption (λmax). Obtain between three and five replicate readings for each standard to assess precision [14].
- Measure Unknowns: Transfer the unknown samples to a cuvette and measure their absorbance under the exact same conditions. Ensure the unknown samples are prepared in the same buffer and at the same pH as the standards [14].
Step 4: Plot the Data and Generate the Calibration Curve
- Plot the average absorbance (y-axis) against the known concentration (x-axis) for each standard.
- Fit the data to a linear regression model (y = mx + b) using statistical software. The output provides the slope (m) and y-intercept (b) of the calibration line [14].
- Examine the plot. A well-behaved curve will appear linear over a specific range, with a non-linear section (limit of linearity, LOL) indicating detector saturation at high concentrations [14].
Step 5: Examine and Validate the Calibration Curve
- Assess Linearity: The coefficient of determination (R²) quantifies the goodness of fit. However, an R² close to 1.0 is not sufficient alone to prove linearity. Use residual plots and other statistical tests (e.g., lack-of-fit test) to validate the model [25].
- Check for Heteroscedasticity: Over a wide concentration range, the variance of the response can change (heteroscedasticity). If the data shows increasing scatter with concentration, a weighted least squares regression should be used to improve accuracy, especially at the lower end of the calibration range [25] [15].
- Use Quality Controls (QC): Quality Control samples prepared at low, medium, and high concentrations within the calibration range should be analyzed to verify the accuracy and precision of the method during validation and routine use [25].
Data Analysis and Advanced Fitting Methodologies
For simple systems, a linear regression is sufficient. However, for complex molecules like conjugated organic dyes, advanced fitting functions may be required to accurately interpret UV-Vis spectra. Recent research demonstrates the efficacy of a modified Pekarian function (PF) for fitting spectra with high accuracy, especially for bands that are vibronically resolved or completely unresolved [26]. This approach, which optimizes five parameters defining the band shape, can provide a more accurate deconvolution of overlapping bands compared to traditional Gaussian or Lorentzian functions, allowing for more precise extraction of quantitative information [26].
Step-by-Step Protocol: Developing Robust UV-Vis Calibration Curves
Ultraviolet-visible (UV-Vis) spectroscopy is a foundational analytical technique in research and drug development, used to determine the concentration of compounds in solution. The principle relies on the measurement of how much ultraviolet or visible light is absorbed by a sample, described by the Beer-Lambert law [2]. This relationship between absorbance and concentration is quantifiable through a calibration curve, making accurate and precise curve construction critical for reliable results. This protocol details the development of a pharmaceutical-compliant UV-Vis calibration curve, providing a standardized methodology for researchers and scientists engaged in compound quantification.
Principles and Theory
In UV-Vis spectroscopy, molecules absorb light of specific wavelengths, promoting electrons to higher energy states. The amount of light absorbed at a given wavelength is directly proportional to the concentration of the analyte in a solution, provided the path length is constant [2]. This relationship is expressed by the Beer-Lambert law:
A = εlc
Where:
- A is the measured Absorbance (no units)
- ε is the molar absorptivity (L·mol⁻¹·cm⁻¹)
- l is the path length of the cuvette (cm)
- c is the concentration of the analyte (mol·L⁻¹)
A calibration curve is a plot of absorbance (y-axis) against known concentrations of standard solutions (x-axis). The data is typically fit with a linear regression, yielding an equation (y = mx + b) that is used to calculate the concentration of unknown samples based on their absorbance [14] [27]. The coefficient of determination (R²) quantifies the goodness of fit, with a value close to 1.0 indicating a highly linear relationship [14].
Materials and Equipment
Research Reagent Solutions and Essential Materials
The following table details the essential materials required for the preparation of standard solutions and execution of the calibration protocol.
Table 1: Essential Materials and Reagents for UV-Vis Calibration Curve Preparation
| Item | Specification / Function |
|---|---|
| Personal Protective Equipment (PPE) | Lab coat, gloves, and safety glasses to ensure user safety [14] [28]. |
| Standard Solution | A concentrated stock solution of the analyte with a known, high purity [14]. |
| Solvent | High-purity solvent (e.g., deionized water, methanol) matching the sample matrix and transparent in the UV-Vis range [14]. |
| Volumetric Flasks | Class A glassware for precise preparation and dilution of standard solutions [29] [30]. |
| Pipettes and Tips | Calibrated precision pipettes and corresponding tips for accurate liquid transfer [14]. |
| Cuvettes | Sample holders; quartz for UV analysis, glass or plastic for visible light only [14] [2]. |
| UV-Vis Spectrophotometer | Instrument comprising a light source, wavelength selector, and detector to measure absorbance [2]. |
| Analytical Balance | For precise weighing of solid solutes to prepare stock solutions [14]. |
| Vortex Mixer | To ensure thorough mixing and homogeneity of solutions (Optional) [14]. |
Instrumentation
A UV-Vis spectrophotometer is the core instrument. Key components include [2]:
- Light Source: Often a combination of a deuterium lamp (UV) and a tungsten or halogen lamp (visible).
- Wavelength Selector: A monochromator with a diffraction grating (typically >1200 grooves/mm) to isolate specific wavelengths.
- Detector: A photomultiplier tube (PMT) or photodiode to convert light intensity into an electrical signal.
For regulated pharmaceutical environments, instruments like the PerkinElmer LAMBDA 365+ with enhanced security (ES) software ensure 21 CFR Part 11 compliance and support workflows from R&D to QC [31].
Experimental Protocol
Preparation of Standard Solutions
- Prepare Stock Solution: Accurately weigh the analyte and transfer it to a volumetric flask. Dilute to the mark with solvent to create a concentrated stock solution [14].
- Perform Serial Dilution: Label a series of volumetric flasks for the standard concentrations.
- Pipette a calculated volume of the stock solution into the first flask.
- Dilute to the mark with solvent and mix thoroughly. This is the first standard.
- Repeat this process, sequentially pipetting from the previous dilution into the next flask and diluting to create a series of at least five standards covering the expected concentration range of the unknown samples [14].
Spectrophotometric Measurement
- Instrument Setup: Turn on the UV-Vis spectrophotometer and allow it to initialize. Set the desired wavelength based on the analyte's absorption maximum [28].
- Establish Baseline: Fill a cuvette with the pure solvent (blank) and place it in the sample holder. Collect a baseline or zero the absorbance [2] [28].
- Measure Standards: Transfer each standard solution into a clean cuvette and place it in the spectrophotometer. Record the absorbance value. Obtain between three and five replicate readings for each standard to assess precision [14].
- Measure Unknowns: Transfer the unknown samples to cuvettes and measure their absorbance following the same procedure used for the standards [14].
Data Analysis and Curve Fitting
- Plot the Data: Create a scatter plot with concentration on the x-axis and the average absorbance for each standard on the y-axis [14].
- Perform Linear Regression: Fit the data to a linear regression model (y = mx + b) using statistical software. The output provides the slope (m, sensitivity) and y-intercept (b) [14] [27].
- Calculate R²: Obtain the coefficient of determination (R²) to evaluate the linearity of the curve. An R² value >0.995 is typically desirable for quantitative work [14].
- Determine Unknown Concentrations: Use the regression equation (y = mx + b) to calculate the concentration (x) of unknown samples based on their measured absorbance (y) [27].
The following workflow diagrams the complete experimental process from preparation to analysis.
Workflow for UV-Vis Calibration
Data Interpretation and Quality Control
Evaluating the Calibration Curve
A well-constructed calibration curve should have a significant linear portion. At higher concentrations, the relationship may become non-linear (limit of linearity), indicating the instrument is nearing saturation [14]. The linear dynamic range should be used for quantification.
Table 2: Key Parameters for Data Analysis and Validation
| Parameter | Description & Target Value |
|---|---|
| Linear Range | The concentration interval over which the absorbance-concentration relationship remains linear. |
| Slope (m) | Represents the sensitivity of the method. A steeper slope indicates higher sensitivity. |
| Y-Intercept (b) | Should be close to zero. A significant offset may indicate matrix interference. |
| Coefficient of Determination (R²) | Quantifies linearity. A value of >0.995 is typically expected for quantitative analysis [14]. |
| Limit of Detection (LOD) | The lowest concentration that can be detected (but not necessarily quantified). |
| Limit of Quantitation (LOQ) | The lowest concentration that can be quantified with acceptable accuracy and precision. |
Quality Assurance and Compliance
For pharmaceutical analysis, compliance with global pharmacopoeia standards (USP, Eur. Ph., JP) is mandatory [31]. This includes:
- Instrument Qualification: Regular operational qualification (OQ) according to standards such as USP <857> to ensure instrument performance [31].
- Volumetric Equipment Calibration: Regular gravimetric calibration of flasks and pipettes is essential for data integrity [29] [30].
- Data Integrity: Use of compliant software with audit trails and electronic records to meet 21 CFR Part 11 regulations [31].
Troubleshooting and Best Practices
- Cuvette Selection: Use quartz cuvettes for UV light measurements, as glass and plastic absorb UV light [2].
- Absorbance Range: Keep absorbance readings below 1.0 to remain within the instrument's dynamic range. Dilute samples that are too concentrated [2].
- Path Length Consistency: Ensure all measurements use cuvettes of the same path length (typically 1 cm) [2].
- Sample Clarity: Ensure samples are clear and free of particulates that could scatter light.
- Temperature Control: Be aware that temperature fluctuations can impact some measurements and reaction kinetics [27].
In quantitative analytical research, particularly in studies involving UV-Visible (UV-Vis) spectroscopy for compound quantification, the preparation of a concentrated stock solution is a fundamental prerequisite for generating reliable calibration curves [32] [33]. A stock solution of accurately known concentration serves as the primary reference from which all subsequent standards and dilutions are derived. The integrity of the entire calibration process, and by extension the accuracy of compound quantification in unknown samples, hinges on the precision and care taken during this initial step [34]. This protocol details a standardized methodology for preparing a concentrated aqueous stock solution, framed within the context of developing a UV-Vis calibration model.
Research Reagent Solutions and Materials
The following table catalogs the essential materials and reagents required for the accurate preparation of a concentrated stock solution.
Table 1: Essential Materials and Reagents for Stock Solution Preparation
| Item Name | Function/Explanation |
|---|---|
| Analytical Balance | Provides precise measurement of solute mass, which is critical for calculating exact molarity [34]. |
| Purified Water | Acts as the solvent; using purified water ensures that impurities do not contaminate the solution or interfere with subsequent UV-Vis analysis [34]. |
| High-Purity Solute | The compound to be quantified (e.g., chalcone, polystyrene nanoplastics). High purity ensures an accurate and specific spectroscopic signal [32] [33]. |
| Volumetric Flask | Designed to contain a precise volume of liquid at a specified temperature, ensuring the final solution volume is accurate [34]. |
| Weigh Boats | Used to contain the solute during weighing on the analytical balance, preventing spillage and contamination [34]. |
| Magnetic Stir Plate & Stir Bar | Facilitates the rapid and even dissolution of the solute in the solvent, leading to a homogeneous stock solution [34]. |
| pH Meter | Monitors and allows for adjustment of the solution's pH, which can be critical for solute stability and UV-Vis absorbance characteristics [34]. |
Experimental Protocol
Calculation of Solute Mass
The first step involves calculating the exact mass of solute required to achieve the desired concentration and volume of the stock solution.
- Define Parameters: Determine the target concentration (e.g., 5 M) and final volume (e.g., 1 L) of your stock solution.
- Obtain Molecular Weight: Find the molecular weight (MW) of the solute, typically listed on the container, in g/mol.
- Apply Formula: Use the following formula to calculate the required mass [34]:
Mass (g) = Concentration (mol/L) × Volume (L) × Molecular Weight (g/mol)Example: To prepare 1 L of a 5 M solution of a compound with a MW of 50 g/mol: Mass (g) = 5 mol/L × 1 L × 50 g/mol = 250 g
Step-by-Step Preparation Procedure
Follow this detailed methodology to prepare the stock solution [34].
- Weigh the Solute: Using an analytical balance and a weigh boat, carefully measure the mass of solute calculated in the previous step.
- Transfer Solvent: Pour approximately three-quarters of the final volume of purified water into a clean beaker. For a 1 L solution, use about 750 mL of water.
- Dissolve the Solute: a. Place a magnetic stir bar into the beaker. b. Set the beaker on a magnetic stir plate and begin stirring. c. Gradually add the weighed solute to the stirring water.
- Adjust pH (if necessary): Once the solute is fully dissolved, measure the pH of the solution. Adjust to the desired pH using dilute sodium hydroxide (to increase pH) or dilute hydrochloric acid (to decrease pH). Add these reagents slowly to avoid overshooting the target.
- Finalize Volume (Q.S. to Volume): a. Using a funnel, quantitatively transfer the solution from the beaker into a volumetric flask of the target volume (e.g., 1 L). b. Carefully add purified water ("q.s." or quantum satis) until the bottom of the meniscus is level with the calibration mark on the neck of the flask. c. Cap the flask and invert it several times to ensure complete mixing and homogeneity.
Workflow and Data Integration for UV-Vis Calibration
The prepared stock solution is the foundation for a workflow that culminates in the creation of a UV-Vis calibration curve, a critical tool for quantifying compounds in unknown samples [32] [33]. This process involves serial dilution, spectroscopic measurement, and data analysis.
Experimental Workflow Diagram
Dilution and Data Generation for Calibration
The concentrated stock solution is used to prepare a series of standard solutions of known concentration through dilution, which are then analyzed via UV-Vis spectroscopy.
Table 2: Example Dilution Scheme and Data Recording for Calibration
| Standard Solution | Volume of Stock (mL) | Volume of Solvent (mL) | Final Concentration (µg/mL) | UV-Vis Absorbance |
|---|---|---|---|---|
| Blank | 0.00 | 10.00 | 0.00 | 0.000 |
| Std 1 | 1.00 | 9.00 | 1.00 | 0.150 |
| Std 2 | 2.00 | 8.00 | 2.00 | 0.305 |
| Std 3 | 4.00 | 6.00 | 4.00 | 0.590 |
| Std 4 | 6.00 | 4.00 | 6.00 | 0.885 |
| Std 5 | 8.00 | 2.00 | 8.00 | 1.180 |
Note: Example assumes a stock solution concentration of 10 µg/mL. The dilution formula used is: C₁V₁ = C₂V₂ [34].
Calibration Curve Construction Logic
The data from Table 2 is used to establish a mathematical relationship between concentration and absorbance, forming the calibration model.
The resulting calibration curve, defined by the equation ( y = mx + c ) (where ( y ) is absorbance, ( m ) is the slope, ( x ) is concentration, and ( c ) is the y-intercept), provides the model to calculate the concentration of an unknown sample based on its measured absorbance [33]. The linearity of this relationship, often indicated by an R² value close to 1 (e.g., 0.9994), validates the method's effectiveness over the specified concentration range [33].
Within the methodology for developing a UV-Vis calibration curve for the precise quantification of analytes in drug development, the preparation of standard solutions via serial dilution is a critical foundational step. The accuracy of the entire analytical procedure hinges on the precision with which these standard concentrations are prepared. This protocol details a reliable method for performing serial dilutions to create a standard curve, enabling researchers to quantify unknown compound concentrations with high confidence [35] [14].
Research Reagent Solutions & Essential Materials
The following reagents and equipment are essential for the accurate preparation of standard solutions and subsequent spectrophotometric analysis [14].
Table: Essential Materials for Serial Dilution and UV-Vis Analysis
| Material/Reagent | Function/Application |
|---|---|
| Concentrated Stock Solution | A solution with a precisely known, high concentration of the analyte of interest. Serves as the source for all subsequent dilutions [35] [14]. |
| Solvent (e.g., Deionized Water, Buffer) | The liquid used to dilute the stock solution. It must be compatible with the analyte and instrument, and should match the solvent of the unknown samples [14]. |
| Pipettes and Disposable Tips | For accurate measurement and transfer of specific, small volumes of liquid, which is crucial for achieving precise dilution factors [14]. |
| Volumetric Flasks or Microtubes | For preparing and containing standard solutions with precise volumes. The choice depends on the required final volume of the standards [14]. |
| UV-Vis Spectrophotometer | The instrument used to measure the absorbance of each standard solution at a specific wavelength, generating the raw data for the calibration curve [14]. |
| Cuvettes | Sample holders that are placed in the spectrophotometer. They must be made of a material (e.g., quartz) compatible with the wavelength range used [14]. |
| Personal Protective Equipment (PPE) | Including gloves, a lab coat, and safety glasses to ensure researcher safety when handling chemical substances [14]. |
Experimental Protocol: Serial Dilution Workflow
This section provides a detailed, step-by-step methodology for preparing standard solutions through serial dilution.
The following diagram outlines the logical sequence of the entire serial dilution and calibration process.
Detailed Step-by-Step Procedure
- Make a Concentrated Stock Solution: Begin by preparing a concentrated stock solution of the standard. Precisely weigh the solute and transfer it to a volumetric flask. Dilute to the mark with the appropriate solvent to achieve a known, high concentration [35] [14].
- Label Dilution Vessels: Label a series of volumetric flasks or microtubes. A minimum of five standards is recommended to establish a reliable calibration curve [35] [14].
- Perform the Serial Dilution:
- Into the first vessel, pipette a specific volume of the concentrated stock solution.
- Change the pipette tip, then add the required volume of solvent to the same vessel. Mix the solution thoroughly to ensure homogeneity. This is your first standard solution [14].
- For the second standard, pipette a volume from the first standard solution into a new vessel containing solvent. Change the tip, mix thoroughly [35].
- Repeat this process sequentially, each time transferring a volume from the previous solution to a new vessel with fresh solvent. This creates a series of standards with progressively lower, known concentrations [14].
- Prepare Samples for Analysis: Transfer each standard solution into clean, labeled cuvettes. Ensure that any unknown samples you wish to analyze are prepared in the same solvent and buffer and at the same pH as the standards to minimize matrix effects [35] [14].
Data Presentation and Calculation
This section summarizes the quantitative data from the dilution series and the subsequent spectrophotometric analysis.
Table: Example Serial Dilution Scheme for a Five-Point Calibration Curve
| Standard Solution | Volume Transferred | Volume of Solvent | Dilution Factor | Concentration (µM) |
|---|---|---|---|---|
| Stock | - | - | - | 100.0 |
| Standard 1 | 5 mL of Stock | 5 mL | 1:2 (2x) | 50.0 |
| Standard 2 | 5 mL of Std 1 | 5 mL | 1:2 (4x) | 25.0 |
| Standard 3 | 5 mL of Std 2 | 5 mL | 1:2 (8x) | 12.5 |
| Standard 4 | 5 mL of Std 3 | 5 mL | 1:2 (16x) | 6.25 |
Table: Expected Absorbance Data and Linear Regression Parameters
| Standard Solution | Concentration (µM) | Mean Absorbance (n=3) | Standard Deviation |
|---|---|---|---|
| Blank | 0.00 | 0.000 | 0.000 |
| Standard 1 | 6.25 | 0.125 | 0.005 |
| Standard 2 | 12.5 | 0.245 | 0.008 |
| Standard 3 | 25.0 | 0.498 | 0.010 |
| Standard 4 | 50.0 | 0.950 | 0.015 |
| Linear Regression | Value | ||
| Slope (m) | 0.019 Abs/µM | ||
| Y-intercept (b) | 0.002 Abs | ||
| Coefficient of Determination (R²) | 0.9998 |
Analysis and Quality Control
Constructing the Calibration Curve
After obtaining the absorbance readings, plot the data with absorbance on the y-axis and concentration on the x-axis. If measured in replicate, calculate and add error bars representing the standard deviation [35]. Use statistical software to fit the data to a linear regression, resulting in the equation y = mx + b, where 'm' is the slope and 'b' is the y-intercept [35] [14].
Evaluating Curve Quality
The coefficient of determination (R²) quantifies the goodness of fit, with a value closer to 1.0 indicating a perfect linear relationship [35] [14]. Visually examine the plot; it should be predominantly linear. A non-linear section at higher concentrations indicates the Limit of Linearity (LOL), a sign that the instrument's detection is nearing saturation [35]. For quantitative analysis, standards should fall within the linear range.
This application note details the critical procedural step of measuring absorbance using an Ultraviolet-Visible (UV-Vis) spectrophotometer, situated within the broader research context of developing precise calibration curves for compound quantification. The accuracy of this measurement step directly determines the reliability of the resulting calibration model, which is fundamental to analytical techniques in drug development, environmental monitoring, and quality control [13]. We outline a standardized protocol, highlight key instrumental parameters, identify potential sources of error with mitigation strategies, and provide guidance for initial data analysis to ensure the generation of high-quality, reproducible data.
Theoretical Foundation
The principle of absorbance measurement is governed by the Beer-Lambert Law, which establishes a linear relationship between the absorbance of a solution and the concentration of the absorbing species [36]. The law is expressed as:
A = εlc
Where:
- A is the measured Absorbance (a unitless quantity) [36]
- ε is the Molar Absorptivity or Absorption Coefficient (L·mol⁻¹·cm⁻¹)
- l is the Path Length of the cuvette (cm)
- c is the Concentration of the analyte (mol·L⁻¹)
This relationship is the foundational principle that allows for the construction of a calibration curve, where absorbance (y-axis) is plotted against known concentrations (x-axis) of standard solutions [13]. The resulting curve provides a mathematical function for determining unknown concentrations of test samples.
Essential Materials and Equipment
The following table catalogues the essential reagents and equipment required for accurate absorbance measurements.
Table 1: Research Reagent Solutions and Essential Materials
| Item | Function and Importance |
|---|---|
| Standard Solution | A solution with a precisely known concentration of the target analyte, used to prepare calibration standards [14]. |
| Compatible Solvent | Dissolves the analyte and standards without absorbing in the measured wavelength range (e.g., deionized water, methanol) [14]. Must be used for all blanks, standards, and samples. |
| UV-Vis Spectrophotometer | The core instrument that emits specific wavelengths of light and measures the intensity of light transmitted through a sample to calculate absorbance [13] [14]. |
| Cuvettes | Sample holders with a defined path length (typically 1 cm). Must be made of material transparent to the wavelengths used (e.g., quartz for UV, glass/plastic for Vis) and be optically clear [14]. |
| Precision Pipettes & Tips | Ensure accurate and precise volumetric transfer of standard and sample solutions during serial dilution and cuvette filling [14]. |
| Volumetric Flasks / Microtubes | Used for the precise preparation and dilution of standard solutions to known concentrations [14]. |
| Personal Protective Equipment (PPE) | Lab coat, gloves, and safety glasses are mandatory for personal protection when handling chemical solutions [14]. |
Detailed Experimental Protocol
Instrument Preparation and Initialization
- Power On: Turn on the spectrophotometer and allow it to initialize for the time specified by the manufacturer (typically 15-30 minutes) to ensure stable lamp output and detector performance.
- Wavelength Selection: Set the instrument to the specific analytical wavelength, which is often the wavelength of maximum absorbance (λmax) for the target compound as determined from a prior spectral scan.
- Blank Measurement: Fill a cuvette with the pure solvent used to prepare your standards and samples. Wipe the outside of the cuvette with a lint-free tissue and place it in the sample compartment. Perform a blank measurement to set the 0.000 Absorbance (100% Transmittance) baseline for the experiment [13].
Sample Measurement and Data Acquisition
- Standard and Sample Loading: Transfer each of your prepared standard solutions and unknown samples into clean, dry cuvettes. Ensure the cuvettes are properly oriented in the compartment with the clear, optical faces in the light path.
- Absorbance Measurement:
- Place the cuvette containing the lowest concentration standard into the sample holder and close the compartment lid.
- Record the absorbance value. For enhanced precision, measure each standard and unknown sample in triplicate [14].
- Between measurements, rinse the cuvette multiple times with the next solution to be measured.
- Data Recording: Systematically record all absorbance values alongside the corresponding standard concentrations or sample identifiers in a laboratory notebook or spreadsheet.
Post-Measurement Workflow
The process of measuring absorbance is part of a larger workflow that culminates in a quantitative calibration model, as summarized below.
Diagram 1: Post-Measurement Workflow
Critical Parameters and Troubleshooting
Key Measurement Considerations
- Path Length Consistency: Ensure all measurements use cuvettes with the same path length. Any variation directly violates the Beer-Lambert Law assumption and introduces error [36].
- Absorbance Range: For optimal linearity and minimum error, maintain absorbance readings between 0.1 and 1.0 [36]. Samples yielding absorbance >1.0 should be diluted to bring them into the linear range of the instrument.
- Stray Light: This is light of unintended wavelengths that reaches the detector, causing non-linearity at high absorbances. It is minimized by regular instrument maintenance and keeping the sample compartment closed during measurement [37].
- Wavelength Accuracy: An incorrectly calibrated wavelength can lead to significant errors in measured absorbance, particularly if measuring on the slope of an absorption peak. Regular calibration of the wavelength scale is essential [37] [38].
Table 2: Common Measurement Errors and Mitigation Strategies
| Error Type | Impact on Measurement | Mitigation Strategy |
|---|---|---|
| Stray Light [37] | Causes negative deviation from the Beer-Lambert law at high absorbance. | Use a well-maintained instrument; ensure compartment is sealed; avoid measuring very high absorbances. |
| Wavelength Inaccuracy [37] [38] | Leads to incorrect absorbance readings, especially on absorption band slopes. | Perform regular wavelength calibration using holmium oxide or deuterium emission lines. |
| Inconsistent Path Length [36] [38] | Directly invalidates the Beer-Lambert relationship (A ∝ l). | Use matched cuvettes; ensure consistent positioning; use instruments with automatic path length correction. |
| Sample Turbidity / Scattering | Falsely increases absorbance due to light loss from scattering. | Centrifuge or filter samples to remove particulates; use a wavelength with less scattering. |
| Instrument Noise [38] | Reduces precision and the ability to measure small absorbance differences. | Allow instrument to warm up sufficiently; use smooth (averaging) functions if available. |
Data Analysis and Initial Interpretation
- Plotting the Calibration Curve: Plot the mean absorbance of each standard solution (y-axis) against its known concentration (x-axis) [14].
- Linear Regression: Fit the data points using a linear least-squares regression to obtain the equation of the line in the form y = mx + b, where y is absorbance, m is the slope, x is concentration, and b is the y-intercept [13] [14].
- Assessing Linearity: Calculate the coefficient of determination (R²). An R² value ≥ 0.999 indicates excellent linearity over the concentration range studied [14] [39]. A lower value suggests potential issues with the standards, instrument, or the linearity of the analyte itself.
- Calculating Unknowns: Use the regression equation to calculate the concentration of unknown samples. Substitute the measured absorbance for y and solve for x (concentration).
By adhering to this detailed protocol and critically evaluating both the measurement process and the resulting data, researchers can ensure that this crucial step contributes to the development of a robust, accurate, and reliable calibration curve for compound quantification.
In the quantification of compounds using UV-Vis spectroscopy, such as in the estimation of total chalcone content or the analysis of nanoplastic suspensions, establishing a reliable calibration curve is a fundamental step [33] [32]. This process involves plotting measured absorbance against known analyte concentrations and performing linear regression analysis to obtain a predictive model. The resulting linear equation, y = mx + b, allows researchers to convert the absorbance of an unknown sample into its concentration accurately and precisely [40]. This document provides detailed application notes and protocols for performing this critical analysis, ensuring data integrity and reproducibility suitable for drug development and other scientific research.
Theoretical Foundation of Linear Regression
Linear regression is a statistical method used to model the relationship between a dependent variable and one or more independent variables by fitting a linear equation to observed data [41]. In the context of a UV-Vis calibration curve, the goal is to find the straight line that best represents the relationship between concentration (the independent variable, X) and absorbance (the dependent variable, Y) [40].
The Linear Regression Model
The simple linear regression model is expressed by the equation: y = mx + b [40] Where:
- y is the predicted value of the dependent variable (Absorbance).
- x is the value of the independent variable (Concentration).
- m is the slope of the line (the estimated change in Y for a 1-unit increase in X). In a calibration context, it represents the sensitivity of the method [40].
- b is the y-intercept (the estimated value of Y when X equals 0) [40].
The Least Squares Method
The "best-fit" line is determined by minimizing the sum of the squares of the vertical distances (residuals) between the observed data points and the predicted values on the line [40] [41]. This is known as the least squares method.
- Residual (Error): The difference between an observed value (yᵢ) and the value predicted by the model (ŷᵢ) [41].
- Objective: Minimize the Sum of Squared Errors (SSE) = Σ(yᵢ - ŷᵢ)² [41].
Experimental Protocol: Developing a UV-Vis Calibration Curve
Materials and Reagents
- Standard Reference Material: High-purity (>95%) analytical standard of the target compound (e.g., chalcone) [33].
- Solvent: Spectroscopic-grade solvent suitable for the analyte (e.g., methanol, ethanol, water). The solvent must be transparent in the UV-Vis wavelength range of interest and not react with the analyte [32].
- Volumetric Flasks: Class A, for precise preparation of stock and standard solutions.
- Micropipettes: Calibrated, for accurate liquid handling.
- UV-Vis Spectrophotometer: Calibrated and qualified.
- Cuvettes: Matched quartz or disposable methacrylate cuvettes, depending on the wavelength range.
Step-by-Step Procedure
- Stock Solution Preparation: Accurately weigh a known mass of the standard reference material. Dissolve and dilute to volume with the appropriate solvent in a volumetric flask to create a stock solution of known, relatively high concentration.
- Standard Solution Preparation: Serially dilute the stock solution to prepare at least five standard solutions spanning a concentration range that is both relevant to the unknown samples and within the linear response range of the instrument. A minimum of five concentration levels is recommended for a reliable calibration [42].
- Spectrophotometric Measurement: a. Zero the spectrophotometer using a cuvette filled only with the solvent (blank). b. For each standard solution, measure the absorbance at the predetermined wavelength of maximum absorption (λmax) for the compound. c. Perform all measurements in triplicate to assess precision.
- Data Tabulation: Record the concentration of each standard and its corresponding average absorbance value in a table.
Table 1: Example Data Table for UV-Vis Calibration of Compound X
| Standard Solution | Concentration (μg/mL) | Absorbance (Average, n=3) |
|---|---|---|
| Blank | 0.00 | 0.000 |
| Std 1 | 2.00 | 0.125 |
| Std 2 | 4.00 | 0.241 |
| Std 3 | 6.00 | 0.378 |
| Std 4 | 8.00 | 0.492 |
| Std 5 | 10.00 | 0.617 |
Data Analysis and Linear Regression
Data Plotting and Model Fitting
- Create a Scatter Plot: Plot the mean absorbance values (Y-axis) against the corresponding concentration values (X-axis).
- Perform Linear Regression: Use statistical software (e.g., GraphPad Prism, R, Python) or a dedicated linear regression calculator to compute the slope (m), y-intercept (b), and the coefficient of determination (R²) for the dataset [40].
- Plot the Regression Line: Overlay the calculated line-of-best-fit (
y = mx + b) onto the scatter plot.
Diagram 1: UV-Vis calibration and regression workflow.
Interpreting the Regression Output
The output of the linear regression analysis provides critical information about the quality of the calibration model [40].
Table 2: Key Linear Regression Outputs and Their Interpretation
| Parameter | Symbol | Interpretation in Calibration Context |
|---|---|---|
| Slope | m | Represents the sensitivity of the method. A higher slope indicates a greater change in absorbance per unit change in concentration. |
| Y-Intercept | b | The theoretical absorbance at zero concentration. Ideally, it should be very close to zero. A significantly non-zero intercept may indicate background interference or a matrix effect [40]. |
| Coefficient of Determination | R² | Quantifies the proportion of variance in the absorbance that can be explained by concentration. An R² value ≥ 0.995 is generally considered excellent for analytical methods [40]. |
| P-value of the Slope | p | Tests the null hypothesis that the slope is zero. A p-value < 0.05 provides strong evidence that a significant linear relationship exists, meaning X (concentration) can be used to predict Y (absorbance) [40]. |
Model Validation and Evaluation Metrics
Beyond R², other metrics help validate the regression model's predictive accuracy [41].
- Mean Squared Error (MSE): The average of the squares of the errors (residuals). It is sensitive to outliers.
MSE = (1/n) * Σ(yᵢ - ŷᵢ)²[41]
- Root Mean Squared Error (RMSE): The square root of MSE. It is in the same units as the dependent variable (absorbance), making it more interpretable.
- Mean Absolute Error (MAE): The average of the absolute differences between observed and predicted values. It is less sensitive to outliers than MSE.
MAE = (1/n) * Σ|yᵢ - ŷᵢ|[41]
Table 3: Example Regression Output for a UV-Vis Calibration Curve
| Parameter | Value |
|---|---|
| Regression Equation | y = 0.0612x + 0.005 |
| R-squared (R²) | 0.9991 |
| Slope (m) | 0.0612 |
| Intercept (b) | 0.005 |
| MSE | 0.00015 |
| RMSE | 0.0122 |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for UV-Vis Spectrophotometry
| Item | Function / Purpose |
|---|---|
| Analytical Standard | A substance of known high purity used to prepare calibration standards and validate the analytical method. It serves as the primary reference [33]. |
| Spectroscopic-Grade Solvent | A pure solvent with low UV absorbance, used to dissolve samples and standards without introducing significant background interference [32]. |
| Buffer Solutions | Used to maintain a constant pH during analysis, which is critical for the stability and consistent absorbance of many compounds, especially in drug development. |
| Reference Cuvettes | High-quality, matched cuvettes that ensure pathlength consistency and minimize errors in absorbance measurements. |
Quality Control and Assurance
A robust calibration protocol includes checks to ensure the model's validity over time and its applicability to unknown samples.
Assessing Assumptions of Linear Regression
Linear regression relies on several key assumptions [41]:
- Linearity: The relationship between X and Y is linear.
- Independence: Observations are independent of each other.
- Homoscedasticity: The variance of the residuals is constant across all levels of X.
- Normality: The residuals of the model are approximately normally distributed.
Diagram 2: Key steps for regression model validation.
Using Quality Control (QC) Samples
QC samples of known concentration, prepared independently from the calibration standards, should be analyzed to verify the predictive accuracy of the model. The calculated concentration of the QC samples should fall within an acceptable range (e.g., ±15% of the known value) for the model to be deemed valid.
In the quantification of compounds using UV-Vis spectrophotometry, the calibration curve serves as the fundamental link between instrumental response and analyte concentration. The coefficient of determination, commonly denoted as R², is a statistical parameter frequently used to evaluate the quality of this relationship [14]. While widely utilized across scientific disciplines, a comprehensive understanding of R²'s proper application, interpretation, and limitations is crucial for researchers, scientists, and drug development professionals relying on accurate analytical data.
This application note examines the role of R² in evaluating UV-Vis calibration curves, addressing common misconceptions and outlining rigorous methodological protocols. We explore complementary statistical measures that provide a more complete assessment of calibration curve quality, with a specific focus on applications in pharmaceutical development and compound quantification.
Theoretical Foundations of R²
Statistical Definition and Calculation
The coefficient of determination (R²) quantifies the proportion of variance in the dependent variable (instrument response) that is predictable from the independent variable (concentration) [14]. In calibration terms, it represents how well the regression line approximates the real data points.
Mathematically, R² is derived from the sum of squares of residuals and the total sum of squares:
[ R^2 = 1 - \frac{SS{res}}{SS{tot}} ]
where ( SS{res} ) is the sum of squares of residuals and ( SS{tot} ) is the total sum of squares.
For calibration curves in analytical chemistry, R² is typically calculated during linear regression analysis, with values ranging from 0.0 to 1.0, where 1.0 indicates a perfect fit where all data points lie exactly on the regression line [14].
Common Misconceptions and Limitations
Despite its widespread use, R² has significant limitations that analysts must recognize:
- R² is insensitive to proportional errors: High R² values can mask systematic errors, particularly when calibration ranges are wide [43].
- R² emphasizes high concentration values: In unweighted linear regression, R² disproportionately weights absolute errors at higher concentrations, potentially overlooking significant relative errors at lower concentrations that are critical for detecting trace compounds [43].
- R² does not indicate appropriateness of model: A high R² value does not necessarily validate the chosen regression model, as non-linear relationships may still produce deceptively high R² values [43].
- R² values are context-dependent: Acceptable R² thresholds vary based on the analytical technique, concentration range, and application requirements [15].
These limitations highlight why R² alone is insufficient for comprehensive calibration curve evaluation, particularly in regulated environments like drug development where accurate quantification is essential.
Complementary Metrics for Curve Evaluation
Relative Standard Deviation (RSD) and Relative Standard Error (RSE)
For average response factor calibrations, the relative standard deviation (RSD) provides a more meaningful measure of calibration quality than R². RSD calculates the standard deviation of calibration factors divided by their mean, typically with acceptance criteria of <15% or <20% [43].
Relative standard error (RSE) extends this concept to regression-based calibrations, calculated from the relative errors at each calibration point [43]. The RSE formula is:
[ RSE = \sqrt{\frac{\sum\left(\frac{x' - x}{x}\right)^2}{d.f.}} ]
where ( x' ) is the back-calculated concentration, ( x ) is the known concentration, and ( d.f. ) represents degrees of freedom.
RSE is particularly valuable because it can be applied to various calibration types and provides consistent evaluation criteria across different regression approaches [43].
Comprehensive Calibration Validation Parameters
A robust calibration evaluation incorporates multiple statistical parameters alongside R², as outlined in Table 1.
Table 1: Key Parameters for Comprehensive Calibration Curve Assessment
| Parameter | Calculation | Acceptance Criteria | Application Context |
|---|---|---|---|
| Coefficient of Determination (R²) | ( 1 - \frac{SS{res}}{SS{tot}} ) | Typically >0.990 or >0.995 | Initial assessment of linear relationship |
| Relative Standard Error (RSE) | ( \sqrt{\frac{\sum\left(\frac{x' - x}{x}\right)^2}{d.f.}} ) | <15% or <20% | Evaluates relative error across concentration range |
| Limit of Detection (LOD) | ( 3.3 \times \frac{\sigma}{S} ) | Signal-to-noise ratio ≥ 3:1 | Determines lowest detectable analyte level |
| Limit of Quantification (LOQ) | ( 10 \times \frac{\sigma}{S} ) | Signal-to-noise ratio ≥ 10:1 | Determines lowest quantifiable analyte level |
| Accuracy (% Recovery) | ( \frac{\text{Measured Value}}{\text{True Value}} \times 100\% ) | 85-115% (context dependent) | Assesses systematic error |
| Precision (% RSD) | ( \frac{\text{Standard Deviation}}{\text{Mean}} \times 100\% ) | <15% or <20% | Evaluates random error |
The parameters in Table 1 provide complementary information about different aspects of calibration quality, with R² representing just one component of a comprehensive validation strategy.
Experimental Protocol for UV-Vis Calibration
Reagent and Material Preparation
Table 2: Essential Research Reagent Solutions for UV-Vis Calibration
| Reagent/Material | Specifications | Function/Role in Analysis |
|---|---|---|
| Primary Standard | High purity (≥99%), known stoichiometry, stable | Provides known analyte concentration for calibration standards |
| Solvent | UV-transparent, HPLC grade if needed, compatible with analyte and cuvette material | Dissolves analyte without interfering absorbance in measured range |
| Volumetric Flasks | Class A, appropriate volumes (e.g., 10, 25, 50, 100 mL) | Precise preparation of standard solutions |
| Pipettes | Calibrated, appropriate volume range with certified tips | Accurate transfer of solutions during serial dilution |
| Cuvettes | Quartz for UV range, optical glass for visible range, matched pathlength | Sample holder with defined pathlength for absorbance measurement |
| UV-Vis Spectrophotometer | Double-beam preferred, validated wavelength and photometric accuracy | Measures light absorbance by samples at specific wavelengths |
Step-by-Step Calibration Procedure
Stock Solution Preparation: Accurately weigh the primary standard and dissolve in appropriate solvent to create a concentrated stock solution of known concentration [14].
Serial Dilution: Prepare a series of standard solutions spanning the expected concentration range of unknown samples. A minimum of five calibration standards is recommended, with concentrations evenly distributed across the range [14].
Spectrophotometric Measurement:
- Set the spectrophotometer to the optimal wavelength for the analyte (determined from preliminary scans).
- Blank the instrument with the solvent used for standard preparation.
- Measure the absorbance of each standard solution in triplicate to assess precision [14].
- Maintain consistent measurement conditions (temperature, time, cuvette orientation).
Data Collection and Recording:
- Record absorbance values for each standard concentration.
- Calculate mean absorbance values for triplicate measurements.
- Note any visual deviations from linearity at high concentrations (indicating detector saturation) [14].
Figure 1: UV-Vis Calibration Curve Development Workflow
Data Analysis and Curve Fitting Strategies
Regression Modeling and Weighting Considerations
The choice of regression model significantly impacts calibration quality assessment:
- Unweighted Linear Regression: Minimizes absolute residuals, but may produce unacceptably large relative errors at low concentrations [43].
- Weighted Regression: Applying 1/(concentration)² weighting ensures relative errors across the calibration range have equal impact, which is typically more appropriate for analytical applications [43].
- Forced Through Zero: May be appropriate when theoretical and empirical evidence support zero response at zero concentration.
The selection of weighting factors should be based on the heteroscedasticity assessment of the calibration data [15]. Analytical methods should specify the regression model, weighting scheme, and acceptance criteria for all validation parameters.
Case Study: Potassium Bromate Determination by UV-Vis
A recent study developing a green UV-Vis method for potassium bromate detection in bread using promethazine demonstrated appropriate application of R² alongside complementary validation parameters [8] [7]. The method reported:
- Linear Range: 0.370-2.570 μg/mL
- R² Value: 0.9962
- LOD: 0.005 μg/g
- LOQ: 0.016 μg/g
- Recovery: 82.968% to 108.542%
This example illustrates how R² functions as one component of a comprehensive validation approach, rather than a standalone quality indicator.
Figure 2: Comprehensive Calibration Curve Evaluation Strategy
Regulatory Considerations and Method Validation
In regulated environments like pharmaceutical development, calibration practices must align with regulatory guidelines from agencies such as the FDA and EMA [15]. These guidelines often specify requirements for:
- Number of calibration standards and replicates
- Calibration range relative to expected sample concentrations
- Acceptance criteria for multiple validation parameters
- Procedures for calibration curve evaluation and documentation
While specific R² thresholds are not always mandated, typical acceptance criteria range from >0.990 to >0.999 depending on the application and analytical technique [15]. However, regulatory focus has shifted toward comprehensive method validation rather than reliance on single parameters like R².
The coefficient of determination (R²) provides valuable initial information about the linear relationship between concentration and instrument response in UV-Vis calibration curves. However, R² alone is insufficient for comprehensive calibration quality assessment. Analytical scientists should adopt a multifaceted evaluation strategy incorporating relative standard error (RSE), residual analysis, accuracy and precision measurements, and detection capability parameters.
This holistic approach ensures reliable quantification results, particularly in critical applications such as drug development where analytical data directly impacts product quality and patient safety. By understanding both the utility and limitations of R², researchers can implement more robust calibration practices that withstand scientific and regulatory scrutiny.
Ultraviolet-visible (UV-Vis) spectroscopy is a fundamental analytical technique used to determine the concentration of compounds in solution based on their absorption of light in the ultraviolet and visible regions of the electromagnetic spectrum (typically 200-800 nm) [18] [2]. The foundation of quantitative analysis using this technique is the calibration curve, which establishes a mathematical relationship between the instrument's response (absorbance) and the analyte concentration [14]. While the Beer-Lambert Law suggests a simple linear relationship (A = εbc) for ideal conditions at low concentrations, many real-world analytical scenarios exhibit non-linear behavior that requires more sophisticated modeling approaches [18] [2].
Quadratic (second-degree polynomial) calibration models provide an essential extension beyond simple linear regression, offering improved accuracy for concentration determinations when the analytical response deviates from perfect linearity. These models are particularly valuable when working with complex samples, higher concentration ranges, or in situations where chemical interactions or instrumental factors create curved response patterns [44]. This application note explores the theoretical foundation, practical implementation, and methodological considerations for quadratic calibration models within the context of developing robust quantification methods for pharmaceutical research and drug development.
Theoretical Foundation
Mathematical Formulation of Quadratic Models
The quadratic calibration model extends the simple linear equation by incorporating a second-order term, resulting in the following mathematical form:
Y = a + bX + cX² + ε [44]
Where:
- Y represents the instrument response (absorbance in UV-Vis spectroscopy)
- X represents the known concentration of the standard
- a is the y-intercept term
- b is the linear coefficient
- c is the quadratic coefficient
- ε represents the random measurement error
This model can capture curvature in the calibration data that would be poorly described by a simple straight line, particularly at higher concentrations where the Beer-Lambert Law may begin to break down due to factors such as chemical associations, electrostatic interactions, or instrumental deviations [44].
Comparison of Calibration Models
Table 1: Characteristics of Different Calibration Models Used in UV-Vis Spectroscopy
| Model Type | Mathematical Form | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Linear [45] | Y = a + bX | Ideal dilute solutions, limited concentration range | Simple computation, easy uncertainty analysis, theoretical basis from Beer-Lambert Law [44] | Limited dynamic range, poor fit for curved data |
| Quadratic [44] | Y = a + bX + cX² | Extended concentration ranges, complex matrices | Captures curvature, broader dynamic range, improved fit for non-ideal systems | Requires more standards, complex uncertainty analysis [44] |
| Power [44] | Y = aXᵇε | Dosage measurements, irradiated materials | Can be linearized via log transformation | Limited to specific response patterns |
| Exponential [44] | Y = e⁻ᵃˣ/(b + cX) | Ultrasonic response, specialized applications | Theoretical basis for certain phenomena | Severe computational challenges |
Experimental Protocol for Quadratic Calibration
Materials and Equipment
Table 2: Essential Research Reagents and Equipment for UV-Vis Calibration Curve Development
| Item | Specifications | Function/Purpose |
|---|---|---|
| Primary Standard | High-purity analyte (>99.5%) | Provides known reference material for accurate standard preparation |
| Solvent | Spectroscopic grade, compatible with analyte and cuvette material [14] | Dissolves analyte without contributing significant background absorption |
| Volumetric Flasks | Class A, appropriate volumes (e.g., 10mL, 25mL, 50mL) [14] | Precise preparation of standard solutions with accurate volumes |
| Pipettes | Calibrated, appropriate volume range with disposable tips [14] | Accurate transfer of specific solution volumes during dilution series |
| UV-Vis Spectrophotometer | With deuterium and tungsten lamps, monochromator, and detector (PMT, photodiode, or CCD) [2] | Measures light absorption at specific wavelengths |
| Cuvettes | Quartz for UV range (200-400 nm), optical glass or plastic for visible range [14] [2] | Sample holders with defined path length (typically 1 cm) |
| Computer with Software | Capable of statistical analysis and regression modeling [14] | Data collection, curve fitting, and concentration calculations |
Step-by-Step Procedure
Step 1: Preparation of Stock Standard Solution
- Precisely weigh an appropriate amount of high-purity reference standard using an analytical balance [14].
- Quantitatively transfer the standard to a volumetric flask and dilute to volume with suitable solvent.
- Calculate the exact concentration of the stock solution. This solution should be at the highest concentration expected in the calibration range.
Step 2: Preparation of Calibration Standards
- Prepare a minimum of five standard solutions spanning the expected concentration range of unknown samples [18] [14]. For quadratic models, additional standards (6-8) are recommended to adequately characterize the curvature.
- Use serial dilution techniques to prepare standards from the stock solution [14]. Pipette the required volume of standard into volumetric flasks and dilute with solvent.
- Ensure standards are prepared in the same matrix as unknown samples to minimize matrix effects.
Step 3: Instrument Measurement
- Zero the spectrophotometer with a blank solution containing all components except the analyte [18] [2].
- Measure the absorbance of each standard solution at the predetermined analytical wavelength.
- Obtain multiple readings (3-5) for each standard to assess measurement precision [14].
- Maintain consistent experimental conditions (temperature, time, cuvette orientation) throughout measurements.
Step 4: Data Analysis and Curve Fitting
- Calculate the average absorbance for each concentration level.
- Plot absorbance (Y-axis) versus concentration (X-axis) to visualize the data trend.
- Perform regression analysis using statistical software to fit the data to a quadratic model (Y = a + bX + cX²).
- Evaluate the goodness of fit using the coefficient of determination (R²) and examination of residuals [14].
Step 5: Model Validation
- Assess whether the quadratic term (c) is statistically significant (p < 0.05).
- Verify that the model adequately describes the data throughout the concentration range.
- Test the model with validation standards not used in curve construction.
- Determine the limit of quantification (LOQ) and limit of detection (LOD) based on the calibrated range.
The following diagram illustrates the decision-making workflow for selecting and implementing an appropriate calibration model:
Data Analysis and Computational Methods
Implementing Quadratic Regression
The process of fitting a quadratic model to calibration data can be implemented through sequential matching, which provides a computationally efficient algorithm that doesn't require complex matrix operations [46]. This approach involves:
- Matching all variables to the constant term: Creating residuals for x, x², and y after accounting for the mean.
- Matching residuals to the linear term: Adjusting for the linear component of the relationship.
- Matching remaining residuals to the quadratic term: Finally capturing the curvature in the data.
The computational implementation can be represented in the following workflow:
Model Evaluation Criteria
When implementing quadratic calibration models, several statistical measures should be examined to ensure model adequacy:
- Coefficient of determination (R²): Quantifies the proportion of variance in the absorbance data explained by the model. Values closer to 1.0 indicate better fit [14].
- Significance of the quadratic term: The p-value for the quadratic coefficient (c) should be statistically significant (typically p < 0.05) to justify the use of a quadratic model over a simpler linear model.
- Analysis of residuals: Residuals (differences between observed and predicted values) should be randomly distributed without systematic patterns.
- Standard error of estimate: Measures the average distance that the observed values fall from the regression line.
Table 3: Example Quadratic Calibration Data for Theoretical Compound X-123
| Concentration (μg/mL) | Average Absorbance | Linear Prediction | Quadratic Prediction | Linear Residual | Quadratic Residual |
|---|---|---|---|---|---|
| 5.0 | 0.125 | 0.118 | 0.127 | +0.007 | -0.002 |
| 10.0 | 0.245 | 0.236 | 0.243 | +0.009 | +0.002 |
| 15.0 | 0.342 | 0.354 | 0.345 | -0.012 | -0.003 |
| 20.0 | 0.438 | 0.472 | 0.435 | -0.034 | +0.003 |
| 25.0 | 0.512 | 0.590 | 0.515 | -0.078 | -0.003 |
| 30.0 | 0.580 | 0.708 | 0.582 | -0.128 | -0.002 |
Applications in Pharmaceutical Research
Quadratic calibration models find particular utility in pharmaceutical analysis where accurate quantification is essential across extended concentration ranges. Specific applications include:
- Drug substance assay: Determining active pharmaceutical ingredient (API) concentration in finished dosage forms when excipients cause matrix effects that produce non-linear responses.
- Dissolution testing: Modeling drug release profiles where concentration ranges can vary significantly throughout the test duration.
- Impurity quantification: Accurate measurement of trace impurities that may exist at concentrations spanning several orders of magnitude.
- Bioanalytical method development: Quantifying drugs and metabolites in biological matrices where complex sample components can interfere with linear response.
In a practical research setting, UV-Vis spectrophotometry combined with quadratic calibration has been successfully applied for water quality monitoring in agricultural settings, demonstrating the methodology's robustness for complex sample matrices [47]. Similar principles can be adapted for pharmaceutical quality control, particularly when dealing with complex formulations or natural product extracts containing multiple interfering compounds.
Limitations and Method Validation Considerations
While quadratic models extend the useful calibration range, they introduce additional complexity that requires careful validation:
- Requirement for more standards: Quadratic models need a minimum of 5-6 concentration levels to adequately characterize the curvature, compared to 3-4 for linear models [44].
- Complex uncertainty analysis: Propagation of error calculations becomes more complicated with quadratic models, making uncertainty estimation less straightforward than with linear models [44].
- Reduced robustness: Quadratic models may be more sensitive to outliers or leverage points at the extremes of the concentration range.
- Extrapolation limitations: Prediction outside the calibrated range is particularly risky with quadratic models, as the curvature may lead to unreasonable predictions.
When implementing quadratic calibration models, it's essential to perform comprehensive method validation including determination of accuracy, precision, specificity, and robustness across the intended concentration range. The model should be periodically verified using quality control samples to ensure ongoing performance.
Troubleshooting UV-Vis Calibration: Solving Common Issues and Enhancing Precision
In the quantification of compounds using UV-Vis spectroscopy, the integrity of analytical data is paramount. This process is foundational to research and drug development, where the accuracy of a calibration curve directly influences the reliability of concentration determinations for unknown samples [14]. The calibration curve, a plot of absorbance versus concentration, serves as the primary tool for quantification, but its validity is entirely dependent on the proper preparation and performance of the spectrophotometer [14] [48].
This application note details the essential pre-measurement protocols of lamp warm-up and instrumental calibration. These steps are critical to mitigate systematic error, ensure compliance with regulatory standards such those found in the pharmacopeias (USP, Ph. Eur.), and guarantee the generation of reproducible and scientifically defensible data [48]. A poorly calibrated instrument can lead to inaccurate potency specifications, failed product batches, and costly investigations, underscoring the non-negotiable nature of these preparatory steps [12] [48].
The Critical Role of Pre-Measurement Checks
Linking Instrument Performance to Data Integrity
The relationship between pre-measurement checks and final analytical outcomes is direct and consequential. Instrumental parameters such as wavelength accuracy, photometric accuracy, and stray light are not abstract specifications; they are fundamental variables that influence the linearity of the Beer-Lambert law (A = εcb) in practical application [48]. Wavelength inaccuracy of just a few nanometers can cause significant errors when measuring on the slope of an absorption peak, while excessive stray light causes negative deviation from linearity at higher absorbances, effectively reducing the usable dynamic range [48].
Regular calibration establishes metrological traceability, creating an unbroken chain of comparison to national standards (e.g., NIST). This process provides objective evidence of data integrity, which is a cornerstone of quality management systems like GLP and GMP, and is mandatory for regulatory filings in the pharmaceutical industry [48].
Pre-Measurement Workflow
The following diagram illustrates the logical sequence of essential pre-measurement checks, connecting preparatory steps to primary calibration and subsequent quantification activities.
Protocol 1: Lamp Warm-Up Procedure
Purpose and Rationale
The light source is the origin of the photon beam used for measurement. A stable spectral output from the deuterium (and/or tungsten) lamp is a prerequisite for reliable photometric data. Upon ignition, lamps require a period to reach thermal and electronic equilibrium. During this time, light intensity and energy distribution can fluctuate, leading to drift in absorbance readings and compromised baseline stability [49]. The warm-up protocol ensures the source output has stabilized before critical measurements are taken.
Step-by-Step Protocol
- Preparation: Ensure the sample compartment is empty and closed. Cuvettes should be cleaned with distilled water and lint-free tissue [49].
- Ignition: Switch on the UV-Vis spectrophotometer. This action automatically ignites the deuterium and tungsten-halogen lamps.
- Initiate Warm-Up: Allow the instrument to remain powered on without taking analytical measurements for a minimum of 30 minutes [49]. For instruments that have been inactive for extended periods (e.g., over a weekend), a longer warm-up time of 60 minutes may be necessary for optimal stability.
- Verification (Optional but Recommended): After the warm-up period, perform a quick baseline scan or monitor the absorbance of a blank solution at a fixed wavelength over a few minutes. The absorbance reading should show minimal drift (e.g., < ±0.001 AU per minute), indicating the system is stable.
Protocol 2: Comprehensive Instrument Calibration
Instrument calibration is a multi-parameter process that verifies the spectrophotometer's key performance characteristics against certified reference materials (CRMs). The following table summarizes the core parameters, their importance, and the standards used for verification.
Table 1: Key Calibration Parameters and Standards Overview
| Calibration Parameter | Fundamental Importance | Primary Reference Materials |
|---|---|---|
| Wavelength Accuracy | Ensures the instrument selects and reports the correct wavelength (x-axis accuracy), critical for identifying compounds and measuring at λmax [48]. | Holmium oxide filter, Didymium filter, Holmium oxide solution [49] [48]. |
| Photometric Accuracy | Verifies the instrument's detector correctly measures absorbance (y-axis accuracy), which is directly tied to concentration calculation errors [48]. | Potassium dichromate solution in 0.005 M sulfuric acid [49] [48]. |
| Stray Light | Quantifies unwanted light outside the selected bandwidth, a major source of error that causes negative deviation from the Beer-Lambert law at high absorbances [48]. | Aqueous potassium chloride (for 200 nm cutoff), sodium iodide (for 220 nm cutoff) [48]. |
| Resolution | Assesses the instrument's ability to distinguish between closely spaced spectral peaks, which is vital for analyzing complex mixtures [49]. | Toluene/hexane solution [49]. |
Calibration Protocols
Wavelength Accuracy Calibration
Principle: To verify that the wavelength indicated by the monochromator corresponds to the actual wavelength of light passing through the sample.
Methodology:
- Using a Holmium Oxide Filter: Place the solid holmium oxide glass filter or a sealed solution cell in the light path.
- Scanning: Perform a scan over the appropriate range (e.g., 200-600 nm).
- Peak Identification: Record the wavelengths of characteristic absorption peaks (e.g., 241.0 nm, 279.4 nm, 287.5 nm, 333.7 nm, 360.9 nm, 418.5 nm, 453.2 nm, 536.2 nm). The exact certified values for your specific CRM should be used [49] [48].
- Acceptance Criteria: The deviation between the observed peak wavelengths and the certified values should typically be within ±1 nm [49].
Photometric Accuracy Calibration
Principle: To confirm that the absorbance values reported by the instrument are accurate across its operational range.
Methodology:
- Solution Preparation: Accurately prepare a potassium dichromate (K₂Cr₂O₇) solution in 0.005 M sulfuric acid. The concentration should be chosen to give absorbances at specific levels (e.g., in the range of 0.2 to 0.8 AU) when measured in a 10 mm pathlength cell [49] [48].
- Measurement: Measure the absorbance of this solution at key wavelengths, typically 235 nm and 257 nm.
- Ratio Calculation: Calculate the ratio of the absorbance at A₂₃₅/A₂₅₇.
- Acceptance Criteria: The observed ratio must fall within the specified limits provided with the CRM or by the pharmacopeia. This verifies the linear response of the detector.
Stray Light Check
Principle: To measure the proportion of extraneous light outside the nominal bandwidth that reaches the detector.
Methodology:
- Solution Preparation: Use a prepared CRM solution designed to block all light at the target wavelength. A common standard is a 1.2% w/v Potassium Chloride (KCl) solution in a quartz cuvette for checking stray light at 200 nm [48].
- Measurement: Place the solution in the beam and measure the apparent "absorbance" at the target wavelength (200 nm for KCl).
- Calculation: The stray light is reported as the percentage transmittance (%T) measured. For example, if the instrument reads 0.001 AU, this is equivalent to 99.77% T, meaning 0.23% stray light.
- Acceptance Criteria: The measured transmittance should be less than a specified limit, often corresponding to an absorbance value greater than 3.0 AU (e.g., %T < 0.1%), though specific limits depend on the instrument and regulatory requirements [48].
Resolution Power Check
Principle: To verify the instrument's ability to resolve fine spectral structure.
Methodology:
- Sample Preparation: Use a solution of toluene in hexane (e.g., 0.02% v/v) [49].
- Scanning: Obtain a spectrum over the range of 260 nm to 270 nm.
- Peak Separation: Examine the resulting spectrum for the clear separation of the peak at 269 nm from the shoulder at 266 nm.
- Acceptance Criteria: The spectrum should show a distinct valley between the two spectral features, confirming the instrument's resolving power.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table catalogs the key materials and reagents required to perform the pre-measurement checks and calibration procedures described in this note.
Table 2: Essential Materials and Reagents for UV-Vis Calibration
| Item | Function / Purpose | Key Specifications |
|---|---|---|
| Certified Reference Materials (CRMs) | Provide traceable standards with known properties for verifying wavelength and photometric accuracy [48]. | Must be NIST-traceable. Examples: Holmium Oxide filter/solution, Potassium Dichromate. |
| Potassium Chloride (KCl) | Aqueous solution used for critical stray light verification in the low UV region [48]. | High Purity (e.g., ACS grade), prepared at 1.2% w/v for 200 nm check. |
| Quartz Cuvettes | Sample holders for UV range measurements; must be transparent down to 190 nm [14] [49]. | High-quality quartz (silica), clean and scratch-free. |
| Volumetric Flasks | For precise preparation and dilution of standard solutions and samples [14]. | Class A glassware to ensure accurate volume measurements. |
| Precision Pipettes and Tips | For accurate and reproducible transfer of liquid volumes during solution preparation [14]. | Calibrated regularly; use tips designed for the specific pipette model. |
| UV-Vis Spectrophotometer | The core analytical instrument that measures the absorption of light by samples [14] [12]. | Equipped with deuterium and tungsten lamps, a monochromator, and a detector. |
Concluding Remarks: Integration with Calibration Curve Development
The rigorous application of lamp warm-up and instrument calibration protocols is the foundation upon which a valid calibration curve is built. Only after confirming the spectrophotometer's performance within specified tolerances can one proceed with confidence to the preparation of standard solutions and measurement of their absorbances for curve construction [14].
A calibration curve developed on a poorly calibrated instrument is inherently unreliable, potentially leading to significant errors in the quantification of research compounds or active pharmaceutical ingredients (APIs) [50] [12]. These pre-measurement checks are therefore not isolated tasks but are integral, non-negotiable first steps in the overarching workflow of quantitative analysis via UV-Vis spectroscopy, ensuring that the resulting data meets the stringent demands of scientific research and drug development.
In the quantification of compounds via UV-Vis spectrophotometry, the calibration curve serves as the fundamental link between instrumental response (absorbance) and analyte concentration. This relationship is governed by the Beer-Lambert Law, which states that absorbance is directly proportional to concentration for a given path length and molar absorptivity [51]. However, this linear relationship holds true only within a specific concentration range. Beyond a certain point, known as the Limit of Linearity (LOL), the response curve deviates from linearity and eventually reaches a plateau where the instrument becomes saturated [14]. This phenomenon represents a critical methodological boundary in analytical chemistry, particularly in pharmaceutical development where accurate quantification is essential for drug characterization, formulation analysis, and quality control. Understanding, identifying, and addressing this non-linear behavior is therefore paramount for developing robust analytical methods that generate reliable data across the required concentration ranges.
Theoretical Foundations: Why Non-Linearity Occurs
Fundamental Principles and Limitations of the Beer-Lambert Law
The Beer-Lambert Law provides the theoretical foundation for UV-Vis spectrophotometry, expressing that the absorbance (A) of a solution is equal to the product of its molar absorptivity (ε), path length (l), and concentration (c): A = εlc [51]. This relationship assumes that: (1) the incident light is monochromatic, (2) the absorbing species act independently of one another, (3) the absorption occurs in a uniform medium, and (4) the sample is homogeneous and does not scatter light. Violations of these underlying assumptions, particularly at higher concentrations, are the primary causes of non-linearity. The logarithmic nature of absorbance, defined as A = log₁₀(I₀/I) where I₀ is the incident light intensity and I is the transmitted light intensity, means that at very high absorbances (typically >2), the transmitted light becomes too weak for the detector to measure accurately, leading to saturation [51].
Key Factors Leading to Saturation and LOL
Several interconnected chemical, physical, and instrumental factors contribute to the deviation from linearity:
- Molecular Interactions: At high concentrations (typically >10⁻² M), the average distance between absorbing molecules decreases significantly. This proximity can lead to electrostatic interactions such as dimerization or aggregation that alter the absorption characteristics of the molecules. These molecular associations can shift absorption maxima or change molar absorptivity, directly violating the assumption that absorbers act independently [51].
- Instrumental Limitations: Every spectrophotometer has a finite dynamic range of detection. As absorbance increases, the intensity of transmitted light (I) decreases exponentially. Eventually, I becomes so small that it approaches the baseline noise level of the detector, making accurate measurement impossible. This instrumental saturation manifests as a flattening of the calibration curve [14] [51].
- Chemical and Matrix Effects: Stray light, scattering in turbid samples, refractive index changes at high concentrations, and chemical equilibria (such as acid-base or association equilibria) that are concentration-dependent can all contribute to non-linearity. The matrix effect can be responsible for either elemental suppression or enhancement, further complicating the concentration-response relationship [52].
Experimental Identification of the Limit of Linearity
Protocol for Assessing Linear Range and LOL
Objective: To empirically determine the linear working range and identify the Limit of Linearity for a compound of interest.
Materials and Equipment:
- UV-Vis spectrophotometer (e.g., Shimadzu UV-2600i) [53]
- Quartz cuvettes (for UV measurements) [14]
- Analytical balance [14]
- Precision pipettes and tips [14]
- Volumetric flasks [14]
- Stock solution of the analyte in appropriate solvent
Procedure:
- Prepare Stock Solution: Accurately weigh and prepare a concentrated stock solution of the analyte in the appropriate solvent system (e.g., pH 7.4 phosphate buffer and ethanol in 1:1 ratio) [54].
- Prepare Standard Solutions: Using serial dilution techniques, prepare a minimum of 8-10 standard solutions covering a broad concentration range. Ensure the range extends beyond the expected linear region to adequately characterize the non-linear portion. For example, in curcumin analysis, a range of 2-10 μg/ml was used [54].
- Measure Absorbance: Using the appropriate solvent as a blank, measure the absorbance of each standard solution at the predetermined absorption maximum (λmax) of the analyte. For Nebivolol Hydrochloride, this was 281-284 nm depending on the solvent system [55]. Perform triplicate measurements for each concentration to assess precision.
- Plot Initial Calibration Curve: Create a scatter plot with concentration on the x-axis and mean absorbance on the y-axis. Visually inspect the plot to identify the point where deviation from linearity begins [14].
- Statistical Analysis: Perform linear regression on progressively narrower concentration ranges to identify where the coefficient of determination (R²) begins to significantly decrease, indicating the onset of non-linearity.
The following workflow diagram illustrates the complete experimental process for identifying the Limit of Linearity:
Data Interpretation and Acceptance Criteria
Visual Inspection: Examine the calibration plot for distinct curvature, particularly at higher concentrations. The Limit of Linearity (LOL) is identified as the concentration point where the curve begins to deviate consistently from the linear trend established at lower concentrations [14].
Residuals Plot Analysis: Calculate and plot the residuals (difference between observed and predicted absorbance values from linear regression). In a properly linear range, residuals should be randomly scattered around zero. Systematic patterns in residuals indicate non-linearity.
Percent Relative Error (%RE) Assessment: A more reliable approach involves calculating the percent relative error for back-calculated concentrations [56]. For each standard, calculate: %RE = [(Ccalculated - Cnominal)/Cnominal] × 100 where Ccalculated is the concentration back-calculated from the regression equation and C_nominal is the known concentration. The linear range is validated when all %RE values fall within acceptance criteria (typically ±15% for pharmaceutical applications) [56]. A proposed fitness-for-purpose criterion is %RETh = 2·C^(-0.15) [56].
R² Limitations: While commonly used, the coefficient of determination (R²) is considered "totally unreliable for linearity assessment" alone, as it can appear acceptable even with significant curvature [56]. It should be used in conjunction with other assessment methods.
Critical Parameters and Troubleshooting
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key materials and reagents essential for successful calibration curve development and LOL assessment:
Table 1: Essential Research Reagents and Materials for Calibration Curve Development
| Item | Specification | Function/Purpose | Critical Considerations |
|---|---|---|---|
| Primary Standard | High-purity certified reference material (CRM) [52] | Provides known concentration for accurate calibration | Purity should be ≥95%; should be traceable to certified standards |
| Solvent System | HPLC/spectrophotometric grade [14] | Dissolves analyte without interfering absorbance | Must be transparent at analytical wavelength; should match sample matrix |
| Volumetric Flasks | Class A precision [14] | Precise volume measurement for standard preparation | Calibration should be verified periodically; use same type for all dilutions |
| Pipettes | Calibrated precision pipettes [14] | Accurate transfer of standard solutions | Regular calibration essential; use positive displacement for viscous solutions |
| Cuvettes | Quartz (UV), optical glass (Vis) [14] | Sample holder for absorbance measurement | Pathlength must be consistent; must be transparent at analytical wavelength |
| Buffer Systems | Analytical grade reagents [54] | Controls pH and ionic strength | pH can affect absorption spectrum; must not absorb at analytical wavelength |
Troubleshooting Common Non-Linearity Issues
When non-linearity occurs at lower concentrations than expected, consider these corrective actions:
- Stray Light Verification: Check instrument specifications for stray light characteristics, which can cause negative deviation from Beer-Lambert Law at higher absorbances. Use appropriate cutoff filters to validate performance [51].
- Dilution Verification: Confirm dilution accuracy by preparing standards using different dilution schemes (e.g., independent weighing vs. serial dilution) to identify technique-related errors.
- Chemical Stability Assessment: Evaluate standard stability over time to detect potential degradation that could cause curvature. Prepare fresh standards and measure absorbance at multiple time points.
- Wavelength Verification: Confirm the accuracy of the selected analytical wavelength by scanning standards across a range to ensure measurement at λmax. Shift in λmax with concentration may indicate molecular associations.
- Pathlength Consideration: For highly absorbing compounds, use shorter pathlength cuvettes (e.g., 1 mm instead of 10 mm) to maintain absorbance within the optimal 0.1-1.5 AU range and extend linear dynamic range.
Data Presentation and Statistical Analysis
Quantitative Analysis of Linearity Parameters
The following table summarizes typical linearity parameters and acceptance criteria for a validated UV-Vis analytical method, based on International Conference on Harmonization (ICH) guidelines [54] [55]:
Table 2: Linearity Parameters and Acceptance Criteria for UV-Vis Method Validation
| Parameter | Experimental Results | Acceptance Criteria | Application Example |
|---|---|---|---|
| Linearity Range | 2-10 μg/ml for curcumin [54]; 10-50 μg/ml for Nebivolol HCl [55] | Should cover 50-150% of target concentration | Pharmaceutical formulation analysis [54] [55] |
| Coefficient of Determination (R²) | ≥0.998 [54] | Typically ≥0.995 (with other assessments) [56] | Measures goodness of fit but insufficient alone [56] |
| Y-intercept Bias | ≤2% of target concentration response | Not significantly different from zero (p>0.05) | Indicates potential spectral interference |
| Slope RSD | <2% for replicate curves | Depends on analyte and concentration | Measures method precision across runs |
| LOD (Limit of Detection) | 0.861 μg/ml for curcumin [54] | Typically signal-to-noise 3:1 [57] | Lowest detectable amount |
| LOQ (Limit of Quantification) | 2.872 μg/ml for curcumin [54] | Typically signal-to-noise 10:1 [57] | Lowest quantifiable amount with precision ±15% [57] |
| Back-calculated Concentration Accuracy | 99.79-100.27% recovery for curcumin [54] | Generally 98-102% [56] | Measured as percent recovery |
Advanced Statistical Approaches
For rigorous assessment of linearity, several statistical methods can be employed:
- LOD and LOQ Calculation: Based on the calibration curve, LOD and LOQ can be determined using the formulas: LOD = 3.3σ/S and LOQ = 10σ/S, where σ is the standard deviation of the response and S is the slope of the calibration curve [57]. The standard error from regression analysis can be used as an estimate of σ.
- Lack-of-Fit Test: This statistical test compares the variability of residuals from the regression line to the pure error variability from replicate measurements. A significant lack-of-fit (p<0.05) indicates the linear model is inadequate for the data.
- Response Factor Plot: Plot the response factor (Absorbance/Concentration) against concentration. In the linear range, this plot should appear as a horizontal line with random scatter. A distinct trend (increasing or decreasing) indicates non-linearity.
Method Validation and Regulatory Considerations
In regulated environments such as pharmaceutical development, demonstrating an appropriate linear range is a mandatory component of method validation according to ICH Q2(R1) guidelines [57]. The fitness-for-purpose approach should guide decisions about linearity, considering the intended application of the method [56]. For methods used in proficiency testing (PT) schemes, which assess laboratory performance, the calibration method must be verified under the laboratory's quality management system, often requiring ISO 17025 accreditation for testing laboratories [52].
Proper documentation of linearity assessment should include the raw data, calibration curve plot, residual plot, percent relative error plot, and statistical analysis demonstrating the validated linear range. This documentation is essential for regulatory submissions and laboratory audits, providing evidence that the method is suitable for its intended purpose throughout the defined concentration range.
Diagnosing and Correcting Noisy or Unstable Absorbance Readings
In the quantification of compounds for drug development and research, the accuracy of a UV-Vis calibration curve is paramount. Noisy or unstable absorbance readings pose a significant threat to data integrity, potentially leading to inaccurate concentration determinations, compromised statistical analysis, and unreliable research conclusions. Such instability can stem from instrument malfunctions, suboptimal sample preparation, or inappropriate experimental conditions. This application note provides researchers and scientists with a systematic framework for diagnosing the root causes of baseline noise and reading drift and offers detailed, actionable protocols to correct these issues, ensuring the generation of robust and reliable calibration curves.
Understanding and Diagnosing Signal Noise
Instrumental noise is a primary contributor to absorbance instability and can be categorized based on its behavior and origin. Understanding the type of noise observed is the first step in diagnosing its cause.
Table 1: Categories and Characteristics of Instrumental Noise
| Noise Category | Main Causes | Typical Appearance on Baseline |
|---|---|---|
Constant Noise (s_T = k_1) [58] |
%T readout resolution; Noise from thermal detectors (common in IR spectrometers). | Relative concentration uncertainty is high at both high and low absorbance; minimum uncertainty at A = 0.4343 [58]. |
Proportional Noise (s_T = k_3 T) [58] |
Fluctuations in source intensity; Inconsistent sample cell positioning. | Relative uncertainty is most significant at low absorbances [58]. |
Photon Detector Noise (s_T = k_2 √(T^2 + T)) [58] |
Noise inherent to photon detectors (common in high-quality UV-Vis instruments). | Relative uncertainty is high at low absorbances but smaller and consistent across absorbances from 0.5 to 2.0 [58]. |
A critical, often overlooked, pre-diagnostic step is to establish what constitutes a "normal" or expected signal-to-noise (S/N) ratio for your specific instrument and method. This baseline can be established using the instrument's onboard diagnostics, often by running a noise test with HPLC-grade water flowing through the cell [59]. The S/N ratio is a key metric, with a limit of detection typically defined as a S/N of 3:1 and a limit of quantitation at 10:1 [59].
A Systematic Diagnostic Workflow
Follow the logical troubleshooting pathway below to efficiently identify the source of unstable readings.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Reliable Absorbance Measurements
| Item | Function & Application | Critical Notes |
|---|---|---|
| UV-Vis Calibration Kit [60] | Diagnoses instrument instability, verifying wavelength and photometric accuracy. | A primary diagnostic tool for early detection of instrument drift [60]. |
| Quartz Cuvettes (for UV range) | Holds liquid sample for analysis; quartz is transparent down to ~190 nm. | Glass/plastic cuvettes absorb UV light, causing erroneous results [61]. |
| HPLC-Grade Water | Serves as a pristine blank and solvent for system suitability tests. | Used to measure inherent instrument noise without sample interference [59]. |
| Lint-Free Wipes | For cleaning optical surfaces of cuvettes to remove dust and fingerprints. | Prevents light scattering, a common source of noise and inaccurate readings [61]. |
| Static/In-Line Mixer | Ensures homogeneous mixing of mobile phases in HPLC systems. | Reduces baseline noise caused by improper solvent mixing [59]. |
| Appropriate Buffer & Solvents | Used to prepare blank and sample solutions. | Solvents like methanol absorb strongly at low wavelengths (<210 nm), increasing noise [59]. |
Detailed Experimental Protocols for Diagnosis and Correction
Protocol 1: Instrument Qualification and Lamp Diagnostics
Purpose: To verify the operational status of the spectrophotometer and determine if the light source is the cause of instability. Materials: UV-Vis spectrophotometer, manufacturer's software, UV-Vis Calibration Kit (optional) [60]. Procedure:
- Access Lamp Hours: Navigate through the instrument's software interface to the system status or maintenance menu. Locate and record the total operational hours for the deuterium or xenon lamp [60].
- Assess Lamp Life: Compare the recorded hours against the manufacturer's specifications. Deuterium lamps typically last 1,000–3,000 hours, while xenon lamps are rated for approximately 500 hours. If the lamp is near or beyond its expected lifespan, replacement is the primary recommended action [60].
- Inspect Baseline Stability: Allow the instrument to warm up for 30 minutes. Perform a blank measurement with an appropriate solvent (e.g., HPLC-grade water) and observe the baseline over 10-15 minutes. A healthy instrument should display a stable, low-noise baseline. Significant drift or high-frequency noise at this stage points to an instrument problem [61].
- Advanced Diagnostic (if available): Use a UV-Vis Calibration Kit to run a formal photometric accuracy and wavelength accuracy test. Follow the kit-specific protocol to obtain quantitative data on instrument performance [60].
Protocol 2: Sample and Cuvette Integrity Assessment
Purpose: To eliminate sample-related artifacts and cuvette imperfections as sources of noise. Materials: Spectrophotometer, matched quartz cuvettes, lint-free wipes, sample and blank solutions [61]. Procedure:
- Cuvette Handling and Cleaning:
- Always handle cuvettes by the frosted or ribbed sides to avoid depositing fingerprints on the optical windows.
- Before measurement, thoroughly wipe all four clear optical faces with a lint-free wipe [61].
- Visually inspect the cuvette against a light source for any scratches, cracks, or residual residue. Scratches scatter light and contribute significantly to noise.
- Cuvette Matching and Orientation:
- For high-precision work, use the exact same cuvette for both the blank and sample measurements.
- If using different cuvettes, ensure they are a matched pair. Verify this by filling both with blank solution and confirming the absorbance reading is nearly identical.
- Always place the cuvette into the holder in the same orientation (e.g., marked side facing forward) to ensure consistent light path properties [61].
- Sample Preparation:
- Ensure your sample is homogeneous and well-mixed. Let it sit for a moment after mixing to allow any large air bubbles to rise to the surface.
- After filling the cuvette, tap it gently with your finger to dislodge any small air bubbles clinging to the optical surfaces, as these act as powerful light-scattering centers [61].
- Confirm the sample concentration is within the ideal linear range of the instrument (absorbance between 0.1 and 1.0 AU). Overly concentrated samples (A > 1.5) are a common cause of non-linearity and unstable readings [36] [61].
Protocol 3: Implementing Baseline Correction
Purpose: To computationally account for instrument noise and light-scattering particulates in the sample that cause an offset in the overall absorbance [62]. Materials: Spectrophotometer with baseline correction software functionality. Procedure:
- Understand the Principle: Baseline correction works by subtracting the absorbance value at a specific, non-absorbing wavelength from the absorbance values across the entire spectrum of interest. This corrects for vertical offset, as illustrated below.
Figure: Workflow for applying a baseline correction to a sample spectrum.
- Select the Baseline Wavelength:
- The optimal wavelength is one where neither your analyte nor the sample buffer absorbs light [62].
- For nucleic acid/protein analysis (UV range): 340 nm is the modern default [62].
- For assays extending into visible light: 750 nm is commonly used [62].
- The selection can be empirically determined by examining a sample scan and identifying a flat, non-absorbing region.
- Apply the Correction:
- In the instrument's software, access the baseline correction function (often in the application or method settings).
- Input the selected baseline wavelength. The software will automatically perform the subtraction during or after data acquisition [62].
- For methods with significant scattering, more advanced curve-fitting approaches based on Rayleigh-Mie scattering equations may be employed [63].
Data Presentation and Analysis
Table 3: Troubleshooting Guide for Common Absorbance Issues
| Observed Problem | Most Likely Causes | Corrective Actions |
|---|---|---|
| High-Frequency Noise & Spikes | 1. Failing lamp (arcing) [59]. 2. Electrical interference [59]. 3. Bubbles in flow cell (HPLC) [59]. | 1. Replace lamp [60] [59]. 2. Ensure proper grounding; use noise filter. 3. Degas mobile phase thoroughly [59]. |
| Drifting Baseline | 1. Insufficient warm-up time [61]. 2. Lamp warming up [61]. 3. Temperature fluctuations [61]. 4. Column dewetting (HPLC) [59]. | 1. Allow 30 min for instrument stabilization [61]. 2. Ensure stable lab temperature. 3. Equilibrate HPLC column with mobile phase [59]. |
| Cannot Zero / Set 100% T | 1. Dirty cuvette or optics [61]. 2. Old lamp with low output [61]. 3. Light leak in compartment [61]. | 1. Clean cuvette; service instrument. 2. Check lamp hours and replace [60] [61]. 3. Ensure sample compartment lid is closed. |
| Negative Absorbance | 1. Blank is "dirtier" than sample [61]. 2. Different cuvettes used for blank and sample [61]. | 1. Re-prepare blank solution. 2. Use the same cuvette for blank and sample [61]. |
| High Noise at Low Wavelengths | 1. Solvent absorption cut-off (e.g., MeOH <205 nm) [59]. 2. High absorbance from buffers [59]. | 1. Use acetonitrile instead of methanol [59]. 2. Use UV-transparent buffers at low wavelengths. |
Optimizing the Working Range and Handling Low-Concentration Inaccuracies
In quantitative analysis using Ultraviolet-Visible (UV-Vis) spectroscopy, the relationship between the concentration of an analyte and its absorbance of light is fundamental. This relationship, described by the Beer-Lambert Law, enables researchers to quantify compounds critical to drug development and pharmaceutical research [64]. The accuracy of this quantification is entirely dependent on the quality and appropriateness of the calibration curve used [65]. A poorly constructed calibration can lead to significant inaccuracies, particularly at the extremes of the working range, compromising research findings and subsequent development decisions.
This application note addresses two interconnected challenges: establishing an optimal working range for UV-Vis calibration curves and mitigating inaccuracies at low concentrations. We provide detailed protocols and evidence-based strategies to enhance the reliability of your quantitative analyses, framed within the context of rigorous scientific practice essential for drug development.
Core Principles of Calibration Curve Optimization
The Beer-Lambert Law and its Limitations
The Beer-Lambert Law establishes that the absorbance (A) of a solution is directly proportional to the concentration (C) of the absorbing species and the path length (b) of the light through the solution: A = εbC, where ε is the molar absorptivity coefficient [64]. This linear relationship forms the theoretical basis for quantitative UV-Vis spectroscopy. However, this relationship is reliable only within a specific concentration range. Deviations from linearity can occur at high concentrations due to electrostatic interactions between molecules or instrumental factors like stray light [64]. For greatest accuracy, absorbance readings should ideally fall within the range of 0.1 to 1.0 Absorbance Units (AU) [64].
Impact of Calibration Design on Low-Concentration Accuracy
The design of the calibration curve itself profoundly impacts accuracy, especially at low concentrations. A common misconception is that a wide calibration range with an excellent correlation coefficient (R²) guarantees accuracy across all levels [65]. In reality, high-concentration standards dominate the statistical fit of the regression line. The absolute error (e.g., in counts per second or absorbance units) is typically larger for high-concentration standards. When a best-fit line is drawn through all points, this larger error from the high standards pulls the regression line toward them, often at the expense of accuracy for the low-concentration standards and samples [65]. Consequently, a sample with a true low concentration may be reported with a significant positive bias if calibrated against a curve that includes inappropriately high standards.
Table 1: Key Factors Affecting Calibration Accuracy and Their Impacts
| Factor | Impact on Calibration | Consequence for Low-Level Quantification |
|---|---|---|
| Calibration Range | An overly wide range allows high-standard errors to dominate the regression fit [65]. | Poor accuracy and inflated detection limits for low-concentration samples. |
| Number of Calibration Standards | A higher number of standards improves the mapping of the detector response [15]. | Enhanced accuracy and precision of the regression model across the range. |
| Matrix Effects | Differences in matrix between standards and samples can cause ion suppression/enhancement (MS) or alter nebulization efficiency (ICP) [15] [66]. | Biased results, as the signal from the sample does not correlate correctly with the calibration curve. |
| Pipetting Technique | Improper technique (angle, depth) can dramatically increase volume delivery error [67]. | Increased random error and poor reproducibility, particularly critical for low-volume aliquots. |
Protocols for Establishing an Optimized Working Range
Protocol 1: Defining the Linear Range of an Assay
Purpose: To empirically determine the upper limit of linearity for a given analyte-instrument combination, ensuring all subsequent calibrations operate within a validated linear range.
Materials:
- Stock standard solution of the target analyte
- Appropriate solvent (e.g., HPLC-grade methanol, water)
- Volumetric flasks and pipettes, recently calibrated [67]
- UV-Vis spectrophotometer with matched cuvettes [64]
Method:
- Prepare a High-Concentration Stock: Prepare a standard solution at the maximum concentration expected for the sample type.
- Serially Dilute: Create a series of 8-10 standards via serial dilution, covering a wide range from a negligible concentration up to the high-concentration stock.
- Measure Absorbance: Analyze all standards in random order, measuring the absorbance at the target analyte's λmax [64].
- Data Analysis: Plot the measured absorbance (y-axis) against the nominal concentration (x-axis).
- Determine Linear Range: The linear range is defined as the concentration interval over which the measured concentration (back-calculated from the linear fit of all data) is within ±10% of the nominal concentration for each standard. The highest concentration meeting this criterion is the upper limit of linearity (ULOQ) [65].
Protocol 2: Designing a Calibration Curve for Low-Level Analysis
Purpose: To construct a calibration curve that provides maximum accuracy for samples with concentrations near the method's detection limit.
Materials: As listed in Protocol 1.
Method:
- Define the Target Range: Based on the expected sample concentrations and the required reporting limit, define a narrow calibration range. For example, if samples are expected below 10 ppb and the reporting limit is 0.1 ppb, a suitable range might be 0.5 to 10-20 ppb [65].
- Select Calibration Standards: Use a blank (solvent only) and a minimum of 5-6 non-zero calibration standards within this narrow range. The standards should be spaced appropriately (e.g., geometrically) to evenly cover the interval [15].
- Avoid High Concentrations: Exclude high-concentration standards that are not relevant to the sample set. This prevents their larger absolute errors from dominating the regression and compromising low-end accuracy [65].
- Verify with Quality Controls (QCs): Analyze independent QC samples at low concentrations within the same batch to verify the accuracy of the calibration curve.
The following workflow summarizes the strategic decision process for optimizing your calibration range:
Mitigating Low-Concentration Inaccuracies: Strategies and Protocols
Addressing Sample Preparation and Handling Errors
Accurate sample preparation is paramount. Errors introduced here cannot be corrected later. Key considerations include:
- Pipette Calibration and Technique: Pipettes must be professionally calibrated on a regular schedule. Use the correct pipette for the volume range, avoiding the very low end of a pipette's capacity to minimize relative error. Always hold the pipette perpendicular to the liquid surface and use a consistent plunger pressure [67].
- Solution Homogeneity: Ensure complete mixing of standards and samples using a vortex mixer or sonication. When vortexing, confirm a tiny whirlpool forms, indicating adequate space for mixing. Note that sonication can heat and degrade thermally labile compounds [67].
- Stability and Solubility: Conduct stability studies on prepared standard solutions to define their shelf life. Research analyte chemistry to confirm all compounds are soluble and stable in the chosen solvent at all calibration levels [67].
Managing Instrumental and Matrix Effects
- Cuvette Care: Use optically matched cuvettes. Scratched or dirty cuvettes scatter light, increasing apparent absorbance. Always handle cuvettes by their frosted sides to avoid fingerprints on the optical surfaces [64].
- Solvent Selection: The solvent must not absorb significantly at the analytical wavelength. Solvents like acetone that absorb in the UV range can obscure sample absorption. Solvent polarity can also cause solvatochromic shifts (bathochromic or hypsochromic) in the λmax value [64].
- Matrix Matching: For complex sample matrices, prepare calibration standards in a matrix that mimics the sample as closely as possible. This accounts for matrix effects that can alter the analyte's absorbance characteristics or cause light scattering [66]. In drug development, this might involve using a placebo formulation or simulated biological fluid.
Protocol 3: Standard Addition for Complex Matrices
Purpose: To accurately quantify analyte concentration in samples where the matrix causes significant interference and cannot be easily matched.
Materials:
- Sample aliquot
- High-purity stock standard of the analyte
- Pipettes and volumetric ware
Method:
- Divide the Sample: Split the sample into at least four equal aliquots.
- Spike the Aliquots: Leave one aliquot unspiked. Add known and increasing amounts of the analyte standard to the other aliquots.
- Dilute to Volume: Ensure all aliquots are brought to the same final volume with solvent.
- Measure Absorbance: Analyze all aliquots and plot the measured absorbance (y-axis) against the concentration of the added standard (x-axis).
- Extrapolate Concentration: Extend the best-fit line of the data points until it intersects the x-axis. The absolute value of the x-intercept represents the original concentration of the analyte in the sample [66] [68].
Table 2: Troubleshooting Common Low-Concentration Inaccuracies
| Problem | Potential Cause | Corrective Action |
|---|---|---|
| High signal in calibration blank | Contaminated reagents, dirty cuvettes, or deposition in the sample introduction system [65] [64]. | Use higher purity reagents, thoroughly clean cuvettes, and implement a robust cleaning protocol for the instrument. |
| Poor reproducibility at low levels | Improper pipetting technique, uncalibrated pipettes, or unstable standard solutions [67]. | Verify pipette calibration gravimetrically, retrain on technique, and prepare fresh standard solutions. |
| Non-linear response at low concentrations | Stray light, chemical interactions, or analyte adsorption to container walls [64]. | Verify instrument performance, use a chelating agent if needed, and use silanized vials to prevent adsorption. |
| Negative concentrations for samples | Contaminated blank or matrix effects causing signal suppression not accounted for in the blank subtraction [65] [15]. | Re-prepare the blank using fresh solvents. Consider using the standard addition method to validate results. |
The following workflow outlines a systematic approach to diagnosing and resolving low-concentration inaccuracies:
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Robust UV-Vis Calibration
| Item | Function | Critical Consideration |
|---|---|---|
| High-Purity Reference Standards | Provides the known analyte for creating calibration standards. | Use materials with certified purity and stability from a reputable supplier. Ensure the formulation is stable until the expiry date [67]. |
| ISO-Grade Solvents | To dissolve and dilute standards and samples. | Low UV-cutoff to avoid interference; high purity to prevent contamination that elevates the blank signal [64]. |
| Calibrated Volumetric Equipment | For accurate and precise measurement and transfer of liquids. | Pipettes must be regularly calibrated. Use air-displacement for aqueous, positive-displacement for viscous or volatile liquids [67]. |
| Optically Matched Cuvettes | Hold the sample for analysis in the spectrophotometer. | Must be clean and unscratched. Ensure material (e.g., quartz, glass) is transparent at the wavelengths used [64]. |
| Stable Isotope-Labeled Internal Standard (if applicable) | Added equally to samples and standards to correct for matrix effects and preparation losses. | Must be chemically identical to the analyte but distinguishable by the detector (more common in MS). Compensates for variable sample preparation recovery [15]. |
| Matrix-Matching Components | To create a calibration matrix that mimics the sample. | Critical for complex biological or formulation matrices. Can be a stripped matrix, synthetic fluid, or placebo mixture [15] [66]. |
In analytical chemistry, particularly in the development of UV-Vis calibration curves for compound quantification in pharmaceutical research, the calibration curve serves as the critical link between instrumental response and analyte concentration [69]. For decades, the use of first-order linear equations (y = mx + b) has been the predominant approach for characterizing calibration curves due to their simplicity, straightforward calculation, and ease of linearity estimation [69] [13]. This linear model, embedded in many official analytical standards and validation protocols, provides a seemingly straightforward path for quantifying active pharmaceutical ingredients (APIs) and impurities in drug formulations [69] [70].
However, this conventional linear approach presents significant limitations that can compromise data quality in pharmaceutical research. The phenomenon of curvature can manifest even within designated working ranges, causing systematic errors that linear models cannot capture [69]. When calibration data exhibits curvature, researchers often resort to removing calibration standards to force a more linear fit, thereby wasting valuable data and potentially reducing the effective working range [69]. In reality, for the same calibration points, second-order non-linear calibration often provides superior fit with smaller residuals compared to first-order linear models [69]. The fundamental issue lies not in the mathematics of quadratic equations, but in the lack of standardized criteria for qualitatively characterizing their curvature and performance [69] [71].
The curvature index represents a novel approach to address these limitations by providing quantitative characterization of calibration curve geometry. This parameter, along with complementary curvature angles, enables controlled implementation of non-linear curves while enhancing the precision of pharmaceutical quantification results [69] [71]. By transitioning from a binary "linear-or-not" assessment to a continuous curvature measurement, researchers can make more informed decisions about calibration model selection and objectively compare the performance of different calibration curves.
Theoretical Foundation: From Linear Regression to Curvature Index
Limitations of Traditional Linear Calibration
The conventional reliance on linear calibration models stems from historical convenience rather than scientific superiority. The coefficient of determination (R²) frequently used to validate linearity is actually a measure of goodness-of-fit rather than a true linearity indicator [69]. This misinterpretation often leads researchers to accept suboptimal linear models when their data would be better represented by quadratic functions. Statistical tests like the Mandel test provide some improvement by offering rudimentary classification of calibration curves as either linear or quadratic, but fail to provide qualitative characterization of curvature degree [69].
The fundamental mathematical limitation of linear models becomes particularly apparent at concentration extremes. At low concentration values, linear models can produce substantial relative deviations that are poorly reflected by traditional correlation coefficients [69]. Similarly, at the upper end of calibration ranges, concentration "saturation" phenomena naturally produce curvature that linear models cannot accommodate [72]. By restricting analysis to linear functions, researchers inadvertently introduce systematic errors that propagate through subsequent pharmaceutical quantification results.
Quadratic Calibration Models
Second-degree polynomial equations (y = ax² + bx + c) provide a more flexible mathematical framework for representing the true relationship between analyte concentration and instrumental response [69]. Unlike linear models, quadratic functions can accommodate the natural curvature that occurs due to instrumental saturation, chemical interactions, or other physicochemical phenomena inherent in analytical measurements [69]. The critical requirement for pharmaceutical applications is that the quadratic function must remain monotonic within the working range (no maximum or minimum), ensuring each concentration corresponds to a unique instrumental response [69].
The resistance to implementing quadratic models historically stems not from their mathematical complexity, but from the absence of standardized quality parameters specifically designed for non-linear curves [69] [71]. While R² values can be calculated for quadratic fits, they do not capture the essential geometric property of curvature that distinguishes different quadratic functions from one another. This gap in analytical methodology has limited the widespread adoption of potentially superior quadratic calibration models in regulated pharmaceutical environments.
Curvature Index and Curvature Angles
The curvature index introduces a quantitative framework for characterizing the degree of curvature in calibration functions [69] [71]. This parameter mathematically describes how sharply a calibration curve bends across its concentration range, providing researchers with an objective metric for comparing different calibration curves. The curvature index is complemented by curvature angles, which measure the angular deviation between consecutive segments of the calibration curve at different concentration points [69].
Table 1: Key Parameters for Characterizing Calibration Curve Geometry
| Parameter | Mathematical Definition | Interpretation | Optimal Range |
|---|---|---|---|
| Curvature Index | Quantitative measure of curve bending | Higher values indicate greater curvature; very high values correlate with poor repeatability | Method-dependent; should be minimized while maintaining adequate fit |
| Curvature Angles | Angular deviation between consecutive calibration segments | Increasing values with concentration indicate progressive curvature | Consistent pattern across concentration range |
| Coefficient of Determination (R²) | Proportion of variance explained by model | Measures goodness-of-fit, not linearity | >0.99 for pharmaceutical applications |
| Mandel Test Statistic | F-ratio comparing linear and quadratic models | Determines whether quadratic fit is statistically superior | p<0.05 indicates significant improvement with quadratic model |
The calculation of curvature parameters begins with establishing the second-degree calibration function from experimental data points. From this equation, the curvature can be computed at various concentration points, with the curvature index typically representing an integrated or averaged value across the working range [69]. In practice, larger curvature index values correlate with increased measurement errors due to poor repeatability, enabling researchers to set acceptance criteria for calibration curve quality [69].
Experimental Protocols and Application Notes
Comprehensive Protocol: Implementing Curvature Index in UV-Vis Calibration
This protocol describes the complete procedure for developing and validating UV-Vis calibration curves with curvature index assessment for pharmaceutical compound quantification, specifically adapted for drug substances like tafamidis meglumine [70].
Materials and Reagents
Table 2: Essential Research Reagent Solutions and Materials
| Item | Specification | Function/Purpose |
|---|---|---|
| Standard Solution | Certified reference material of target analyte (e.g., tafamidis meglumine) | Provides known concentration points for calibration curve construction |
| Solvent | HPLC-grade methanol, acetonitrile, or buffer solution appropriate for analyte | Dissolves analyte and creates matrix-matched standards; methanol preferred for green chemistry [70] |
| Volumetric Flasks | Class A, various volumes (10mL, 25mL, 50mL, 100mL) | Precise preparation of standard solutions at specific concentrations |
| Pipettes and Tips | Calibrated variable-volume pipettes (e.g., 100-1000μL, 50-200μL) with compatible tips | Accurate transfer of solution volumes during serial dilution |
| UV-Vis Spectrophotometer | Double-beam instrument with wavelength scanning capability (190-800nm) | Measures absorbance of standards and samples at optimal wavelength |
| Cuvettes | Quartz (UV range) or glass (visible range), 1cm pathlength | Sample holders with consistent light path length for absorbance measurements |
| Software | Microsoft Excel, Origin, or specialized spectrophotometer software | Data collection, regression analysis, and curvature index calculation |
Step-by-Step Procedure
Stock Solution Preparation: Accurately weigh approximately 10mg of reference standard (tafamidis meglumine) and transfer quantitatively to a 10mL volumetric flask. Dissolve with and dilute to volume with appropriate solvent (e.g., methanol) to obtain a final concentration of approximately 1mg/mL [70].
Serial Dilution for Calibration Standards: Perform serial dilutions to prepare a minimum of five standard solutions covering the validated working range (e.g., 3-18μg/mL for tafamidis meglumine) [70]. Use the following scheme:
- Label five volumetric flasks (e.g., 10mL) with respective concentrations.
- Pipette appropriate volumes of stock solution into each flask using calibrated pipettes.
- Dilute to volume with solvent, cap, and mix thoroughly by inversion.
Spectrophotometric Measurement:
- Zero the spectrophotometer with solvent blank at predetermined analytical wavelength.
- Measure absorbance of each standard solution in triplicate, randomizing sequence to minimize systematic error.
- Record average absorbance values for each concentration.
Data Analysis and Model Fitting:
- Input concentration (x) and average absorbance (y) data into statistical software.
- Perform both linear (y = mx + b) and quadratic (y = ax² + bx + c) regression.
- Calculate R² values for both models and perform Mandel test to determine if quadratic fit provides statistically significant improvement.
Curvature Index Calculation:
- From the quadratic equation, calculate the curvature at multiple points across the concentration range.
- Compute the curvature index as defined in Section 2.3.
- Determine curvature angles between consecutive calibration segments.
Validation:
- Prepare and measure independent quality control samples at low, medium, and high concentrations within the calibration range.
- Calculate accuracy (percentage recovery) and precision (%RSD) using both linear and quadratic models.
- Compare performance between models, expecting improved accuracy with quadratic fit for curved data.
Workflow: Implementing Curvature Index in Calibration Strategy
The following workflow diagram illustrates the decision process for implementing curvature index assessment in pharmaceutical calibration development:
Case Study: Pharmaceutical Application with Tafamidis Meglumine
Recent research on tafamidis meglumine quantification demonstrates the practical implementation of advanced calibration approaches in pharmaceutical analysis [70]. Four spectrophotometric methods were developed and validated following ICH guidelines, employing both zero-order and first-order derivative techniques across the concentration range of 3-18μg/mL. All methods exhibited excellent linearity with R² values between 0.9980-0.9995, alongside recovery rates of 99.00%-100.57% and precision values below 2% RSD [70].
This case study exemplifies the modern approach to pharmaceutical method development that incorporates sustainability metrics alongside traditional validation parameters. The use of methanol as a green solvent and evaluation with AGREE and ComplexGAPI metrics demonstrates how comprehensive method assessment extends beyond mere calibration linearity to include environmental impact and practicality considerations [70] [72]. While the published study emphasized linearity, the methodological framework provides an ideal foundation for implementing curvature index assessment to potentially extend the working range or improve accuracy at concentration extremes.
Data Analysis and Interpretation
Comparative Performance: Linear vs. Quadratic Calibration
Robust comparison between linear and quadratic calibration models requires evaluation of multiple performance metrics beyond traditional R² values. The following table summarizes representative data from ion chromatography determination of chloride, illustrating the dramatic improvement achievable with quadratic fitting, particularly at lower concentrations [69]:
Table 3: Comparative Percentage Error (PE) Between Linear and Quadratic Models
| Chloride Concentration (mg/L) | PE with 1st Degree Equation (%) | PE with 2nd Degree Equation (%) | Improvement Factor |
|---|---|---|---|
| 0.1 | 1284.07 | 421.61 | 3.0× |
| 0.5 | 290.77 | 90.06 | 3.2× |
| 1.0 | 132.00 | 39.06 | 3.4× |
| 2.0 | 51.17 | 11.00 | 4.7× |
| 5.0 | 8.04 | 1.76 | 4.6× |
| 10.0 | 2.98 | 0.83 | 3.6× |
The data clearly demonstrates that percentage errors with linear models can be extraordinarily high at lower concentrations, precisely where sensitive detection is often most critical in pharmaceutical analysis [69]. The quadratic model substantially reduces these errors across the entire concentration range, with the most dramatic improvements observed at the lowest concentrations where linear models perform poorest.
Curvature Index in Method Validation
The curvature index serves not only as a descriptive parameter but as a quality control tool for calibration curves. Research across multiple analytical techniques (UV-Vis spectrophotometry, atomic absorption spectrometry, ion chromatography, and catalytic combustion) demonstrates that larger curvature index values correlate strongly with increased measurement errors due to poor repeatability [69]. This relationship enables laboratories to establish validation criteria based on curvature index thresholds, rejecting calibration curves that exceed predetermined curvature limits regardless of their R² values.
Implementation of curvature index assessment revealed that calibration curves typically exhibit increasing curvature angles at higher concentration points, reflecting the progressive deviation from linearity as instrumental response approaches saturation [69]. This pattern holds across different analytical techniques, suggesting a fundamental relationship between concentration magnitude and curvature manifestation that should be incorporated into method development strategies.
Holistic Method Evaluation Framework
Modern analytical method development extends beyond traditional performance parameters to include comprehensive assessment using tools like the RGB model (Red for analytical performance, Green for environmental impact, Blue for practicality) and emerging frameworks such as White Analytical Chemistry [72]. These approaches complement curvature index assessment by providing multidimensional evaluation of method quality.
Recent innovations in method assessment include the Violet Innovation Grade Index (VIGI) which evaluates 10 criteria including sample preparation, instrumentation, data processing, regulatory compliance, and automation [72]. Similarly, the Graphical Layout for Analytical Chemistry Evaluation (GLANCE) condenses complex method descriptions into 12 standardized blocks to enhance communication and reproducibility [72]. These tools represent the evolving landscape of analytical method assessment in which curvature index finds its application as a specialized parameter for calibration quality characterization.
The implementation of curvature index assessment represents a significant advancement in calibration methodology for pharmaceutical analysis. By providing quantitative characterization of calibration curve geometry, this approach enables researchers to move beyond the simplistic linear-versus-quadratic dichotomy to a more nuanced understanding of calibration performance. The demonstrated superiority of quadratic models in reducing percentage errors, particularly at critical low concentration levels, challenges the historical preference for linear fitting in UV-Vis spectrophotometry and other analytical techniques.
Integration of curvature index assessment into routine method validation protocols provides pharmaceutical scientists with a powerful tool for optimizing calibration ranges, improving quantification accuracy, and establishing scientifically justified acceptance criteria. When combined with emerging holistic assessment frameworks like White Analytical Chemistry, curvature index contributes to the development of more reliable, sustainable, and practical analytical methods for drug development and quality control. As the field continues to evolve, the adoption of these advanced calibration strategies will play an increasingly important role in ensuring the accuracy and credibility of pharmaceutical quantification results.
Validation and Technique Comparison: Ensuring Reliability in Complex Matrices
In the development of analytical methods, particularly when using UV-Vis spectrometry for compound quantification, establishing the lowest levels at which an analyte can be reliably detected and measured is a fundamental requirement for method validation. The Limit of Detection (LOD) and Limit of Quantification (LOQ) are two critical performance characteristics that define the sensitivity and utility of an analytical procedure. The LOD represents the lowest concentration of an analyte that can be reliably distinguished from the analytical background noise, but not necessarily quantified as an exact value [73] [74]. In practical terms, it is the minimum concentration that the method can identify as "present" with a defined level of confidence. The LOQ, conversely, is the lowest concentration at which the analyte can not only be detected but also quantified with acceptable precision and accuracy under stated experimental conditions [73] [75]. It represents the lower limit of the quantitative working range of the method.
The accurate determination of these parameters is especially crucial in pharmaceutical research and drug development, where decisions regarding impurity profiling, toxicological assessment, and formulation stability often depend on the reliable measurement of compounds at trace levels. For UV-Vis spectroscopic methods, which are widely employed due to their simplicity, cost-effectiveness, and rapid analysis time, understanding the capabilities and limitations of the method at low concentration levels is essential for ensuring that the technique is "fit for purpose" [73] [76].
Key Definitions and Fundamental Concepts
The Distinction Between LOD and LOQ
A clear understanding of the distinction between LOD and LOQ is paramount for proper method validation. The following table summarizes their core differences:
Table 1: Core Differences Between LOD and LOQ
| Parameter | Limit of Detection (LOD) | Limit of Quantitation (LOQ) |
|---|---|---|
| Definition | The lowest analyte concentration that can be reliably distinguished from the blank [73] [77]. | The lowest concentration that can be quantified with acceptable precision and accuracy [73] [77]. |
| Primary Implication | Detection is feasible [73]. | Reliable quantification is possible [73]. |
| Typical Signal Assurance | The analyte signal is distinguishable from noise with a high degree of confidence [75]. | The signal is sufficient to measure with predefined bias and imprecision goals [73]. |
| Common Applications | Qualitative identification, limit tests for impurities [78]. | Quantitative determination of impurities and low-level compounds [78]. |
In some guidelines, particularly from clinical laboratory sciences (CLSI EP17), a third parameter, the Limit of Blank (LoB), is introduced as a fundamental component for determining LOD [73] [77]. The LoB is defined as the highest apparent analyte concentration expected to be found when replicates of a blank sample (containing no analyte) are tested [73]. It characterizes the background noise and potential false-positive signals of the method. Statistically, it is calculated as the mean blank signal plus 1.645 times its standard deviation (assuming a Gaussian distribution), which establishes a threshold where only 5% of blank measurements would exceed this value due to random noise [73]. The LOD is then determined in relation to the LoB, ensuring that a low-concentration sample can be reliably distinguished from the blank [77].
Methodologies for Calculating LOD and LOQ
There are multiple accepted approaches for determining LOD and LOQ. The choice of method often depends on the nature of the analytical technique, regulatory requirements, and the available data.
Signal-to-Noise Ratio (S/N)
This approach is directly applicable to analytical techniques that display a baseline noise, such as chromatographic or spectroscopic methods [78] [74].
- Principle: The signal from a low-concentration analyte is compared to the background noise level of the system.
- Typical Ratios: An S/N ratio of 2:1 or 3:1 is generally accepted for estimating the LOD [78] [74]. For the LOQ, a ratio of 10:1 is typically required [78] [74].
- Application: This method is straightforward and is often used in chromatographic methods like HPLC [78].
Standard Deviation of the Blank and the Calibration Curve Slope
This is a widely used and statistically rigorous method recommended by guidelines such as ICH Q2(R1) [78] [76] [75].
- Principle: The standard deviation (σ) of the response (either from the blank or from the residual standard deviation of a calibration curve) is used in conjunction with the slope (S) of the calibration curve to estimate the limits.
- Standard Formulas:
- Source of σ (Standard Deviation): The estimate of σ can be derived from:
- The standard deviation of the response for multiple measurements (e.g., n=10-20) of a blank sample [78] [75].
- The residual standard deviation of the regression line (standard error of the y-intercept) from a calibration curve prepared using samples with analyte concentrations in the range of the expected LOD/LOQ [78].
The following diagram illustrates the logical workflow for selecting and applying the most common calculation methods:
Visual Evaluation
This non-instrumental approach involves the analysis of samples with known concentrations of the analyte and establishing the minimum level at which the analyte can be reliably detected or quantified by visual inspection [78] [75]. It is common in techniques such as titrations or inhibition zone tests [78].
Experimental Protocol for UV-Vis Spectrophotometry
This protocol outlines a detailed procedure for determining the LOD and LOQ of a compound using a UV-Vis spectrophotometer, based on the standard deviation of the blank and calibration curve slope method.
Research Reagent Solutions and Materials
Table 2: Essential Materials and Reagents for LOD/LOQ Determination via UV-Vis
| Item | Function / Specification |
|---|---|
| UV-Vis Spectrophotometer | Instrument for measuring light absorbance. Must be qualified and calibrated. |
| Quartz Cuvettes | Sample holders with a defined pathlength (e.g., 1 cm). Must be matched and clean. |
| Analytical Balance | For accurate weighing of the analyte standard (precision ≥ 0.1 mg). |
| Analyte Reference Standard | High-purity compound of known identity and purity. |
| Solvent (e.g., Water, Methanol) | High-purity solvent suitable for UV-Vis analysis, free of interfering absorbances. |
| Volumetric Flasks | Class A glassware for precise preparation of standard solutions. |
| Micropipettes | For accurate and precise transfer of liquid volumes. |
Step-by-Step Procedure
Step 1: Preparation of Standard Stock Solution Accurately weigh about 10 mg of the reference standard. Transfer it quantitatively to a 100 mL volumetric flask, dissolve, and dilute to the mark with the appropriate solvent to obtain a primary stock solution of approximately 100 μg/mL [76].
Step 2: Selection of Analytical Wavelength Dilute an aliquot of the primary stock solution to a concentration of approximately 5-10 μg/mL. Scan this solution over the UV-Vis range (e.g., 200-400 nm) against a solvent blank to identify the wavelength of maximum absorption (λmax) [76].
Step 3: Construction of the Calibration Curve From the primary stock solution, prepare a series of standard solutions covering a suitable range (e.g., 5–30 μg/mL) [76]. Measure the absorbance of each standard solution at the predetermined λmax against the solvent blank. Perform each measurement in triplicate to assess repeatability.
Step 4: Data Analysis and Calculation
- Plot the Calibration Curve: Plot the mean absorbance (y-axis) against the corresponding concentration (x-axis).
- Perform Linear Regression: Calculate the slope (S) and y-intercept of the regression line using statistical software. The correlation coefficient (r) should be >0.995 to demonstrate good linearity [76].
- Calculate LOD and LOQ: Use the residual standard deviation (σ) from the linear regression analysis as the estimate of standard deviation.
- LOD = 3.3 × σ / S
- LOQ = 10 × σ / S For example, in the validation of a UV method for Terbinafine hydrochloride, the LOD and LOQ were found to be 1.30 μg and 0.42 μg, respectively, using this calculation method [76].
Step 5: Experimental Verification (Crucial Step) Once the LOD and LOQ are calculated, it is essential to verify these limits experimentally [74]. Prepare samples at the calculated LOD and LOQ concentrations (at least n=6 for LOQ) and analyze them using the validated method. For the LOQ, the results should demonstrate a precision (expressed as % Relative Standard Deviation, %RSD) of ≤ 20% and an accuracy (expressed as % recovery) of 80-120% [77]. If these criteria are not met, the LOQ should be re-estimated at a slightly higher concentration.
The experimental workflow for this protocol is summarized in the following diagram:
Regulatory Considerations and Best Practices
When validating an analytical method for regulatory submission, it is imperative to follow relevant guidelines such as the International Council for Harmonisation (ICH) Q2(R2) guideline, "Validation of Analytical Procedures" [74] [79]. The methodology for determining LOD and LOQ should be clearly specified in the method validation protocol. Furthermore, the acceptance criteria for precision and bias at the LOQ should be pre-defined based on the intended use of the method [73] [77]. For quantitative impurity tests, the LOQ must be demonstrated to be sufficiently low to control the impurity at or below its specified reporting threshold.
Recent scientific literature indicates a growing use of graphical validation tools, such as the accuracy profile and uncertainty profile, which provide a more holistic assessment of method performance at low concentrations, including the LOQ, by combining tolerance intervals and measurement uncertainty [80]. These strategies are considered a reliable and realistic alternative to classical statistical concepts.
The cosmetic industry continually seeks innovative active ingredients that offer anti-aging benefits with improved stability and reduced side effects compared to traditional retinoids. Bakuchiol, a meroterpene natural product isolated from Psoralea corylifolia seeds, has emerged as a promising retinol-like compound that activates similar gene pathways without associated photodegradation and irritation concerns [81] [82]. As bakuchiol gains prominence in commercial cosmetic formulations, developing robust, efficient analytical methods for its quantification becomes essential for quality control and regulatory compliance.
This case study investigates the application of UV-Vis spectroscopy for quantifying bakuchiol in cosmetic formulations, contextualized within broader research on developing calibration curves for compound quantification. We present a detailed protocol demonstrating how UV-Vis methodology compares with established techniques like HPLC and NMR, highlighting its advantages for routine analysis while acknowledging its limitations in complex matrices [81] [82]. The approach aligns with the cosmetic industry's growing need for accessible analytical techniques that can handle diverse formulation types while maintaining accuracy and precision.
Theoretical Background
UV-Vis Spectroscopy Principles
Ultraviolet-visible (UV-Vis) spectroscopy measures the absorbance of light energy in the ultraviolet (200-400 nm) and visible (400-800 nm) regions of the electromagnetic spectrum. When sample molecules encounter this energy, electrons transition from ground state to excited singlet states, producing characteristic absorption spectra [18]. The fundamental principle governing quantitative analysis is the Beer-Lambert Law:
A = εbc
Where A is absorbance (unitless), ε is the molar absorptivity (M⁻¹cm⁻¹), b is the path length of the cuvette (cm), and c is the concentration (M) [18]. This linear relationship between absorbance and concentration enables the construction of calibration curves for quantifying unknown analytes like bakuchiol in solution.
Bakuchiol Spectroscopic Characteristics
Bakuchiol exhibits strong UV absorption due to its phenolic structure with extended conjugation, featuring a characteristic maximum at λ = 262 nm in ethanol [82]. This distinct absorption peak provides the theoretical basis for selective quantification in cosmetic matrices. The compound's solubility profile—insoluble in water but soluble in alcohols, DMSO, plant oils, and triglyceride/silicone oils—informs appropriate solvent selection for sample preparation and analysis [82].
Experimental Design and Workflow
The following diagram illustrates the complete experimental workflow for bakuchiol quantification in cosmetic formulations using UV-Vis spectroscopy:
Materials and Methods
Research Reagent Solutions
Table 1: Essential Materials and Reagents for Bakuchiol Quantification
| Item | Specification | Function/Importance |
|---|---|---|
| Bakuchiol Standard | High-purity (>95%) reference standard | Provides known concentration for calibration curve [82] |
| Ethanol (Absolute) | HPLC grade or analytical grade | Extraction solvent and dilution medium; bakuchiol shows good solubility [82] |
| Volumetric Flasks | Class A, various sizes (10-100 mL) | Precise preparation of standard solutions and accurate dilutions [14] |
| Micropipettes | Calibrated, various ranges (100-1000 μL) | Accurate transfer of standards and samples [14] |
| UV-Vis Spectrophotometer | Scanning capability 200-800 nm | Instrument for absorbance measurements [18] [14] |
| Quartz Cuvettes | 1 cm pathlength, UV-transparent | Sample holder for UV measurements below 350 nm [14] |
| Centrifuge | Capable of 4000-5000 rpm | Separates insoluble components from cosmetic extracts [82] |
| Syringe Filters | 0.45 μm PTFE or nylon | Removes particulate matter from samples before analysis [83] |
Detailed Experimental Protocols
Calibration Curve Construction Protocol
Step 1: Preparation of Stock Standard Solution
- Accurately weigh 10.0 ± 0.1 mg of pure bakuchiol reference standard
- Transfer quantitatively to a 100 mL volumetric flask using ethanol
- Dilute to mark with ethanol and mix thoroughly to yield 100 μg/mL stock solution
- Store protected from light at 4°C for up to one month [82]
Step 2: Serial Dilution for Standard Solutions
- Prepare a minimum of five standard concentrations spanning the expected sample concentration range
- Recommended concentration series: 2, 5, 10, 15, and 20 μg/mL
- Using volumetric pipettes, transfer appropriate aliquots from stock solution (e.g., 0.2, 0.5, 1.0, 1.5, 2.0 mL) to separate 10 mL volumetric flasks
- Dilute to mark with ethanol and mix thoroughly [14]
Step 3: Spectrophotometric Measurement
- Zero the UV-Vis spectrophotometer with ethanol blank
- Measure absorbance of each standard solution at λ = 262 nm
- Perform triplicate measurements for each concentration to assess precision
- Record average absorbance values for each concentration [14] [13]
Step 4: Calibration Curve Plotting and Validation
- Plot average absorbance (y-axis) versus concentration (x-axis)
- Perform linear regression analysis to obtain equation: y = mx + b
- Calculate coefficient of determination (R²); acceptable values ≥0.990 [14]
- Verify linear range and identify limit of quantification (LOQ)
Table 2: Representative Bakuchiol Calibration Data
| Concentration (μg/mL) | Absorbance 1 | Absorbance 2 | Absorbance 3 | Mean Absorbance | Standard Deviation |
|---|---|---|---|---|---|
| 2.0 | 0.095 | 0.098 | 0.093 | 0.095 | 0.002 |
| 5.0 | 0.231 | 0.228 | 0.235 | 0.231 | 0.004 |
| 10.0 | 0.462 | 0.458 | 0.465 | 0.462 | 0.004 |
| 15.0 | 0.692 | 0.698 | 0.687 | 0.692 | 0.006 |
| 20.0 | 0.920 | 0.925 | 0.918 | 0.921 | 0.004 |
Sample Preparation Protocol for Cosmetic Formulations
The extraction efficiency varies significantly based on formulation type, as illustrated in the following diagram:
For Oil-Based Formulations (Samples 1-4):
- Accurately weigh 100.0 ± 0.1 mg of cosmetic product
- Transfer to 10 mL volumetric flask with ethanol
- Vortex mix for 1 minute until complete dissolution
- Dilute to mark with ethanol; further dilute if necessary to fall within calibration range [82]
For Emulsion-Based Formulations (Samples 5-6):
- Accurately weigh 100.0 ± 0.1 mg of product
- Add 10 mL ethanol and sonicate for 15 minutes
- Centrifuge at 5000 rpm for 10 minutes to separate phases
- Collect clear supernatant for analysis
- Note: Emulsion formulations may require multiple extraction steps for complete recovery [82]
Quantification Protocol
Step 1: Sample Measurement
- Zero spectrophotometer with ethanol blank
- Measure absorbance of prepared samples at λ = 262 nm
- Perform triplicate measurements for each sample extract
Step 2: Concentration Calculation
- Apply calibration curve equation to calculate concentration in sample extract
- Account for all dilution factors in final calculation:
Coriginal = (Cmeasured × Vfinal × DF) / msample
Where Coriginal is bakuchiol concentration in original cosmetic (%, w/w), Cmeasured is concentration from calibration curve (μg/mL), Vfinal is final extract volume (mL), DF is additional dilution factor (if applicable), and msample is original sample mass (mg) [82]
Step 3: Method Validation
- Assess precision via relative standard deviation (%RSD) of replicate measurements
- Determine accuracy through spike recovery experiments
- Calculate method detection limits based on calibration statistics
Results and Discussion
Method Performance and Comparison
Table 3: Comparison of Analytical Methods for Bakuchiol Quantification
| Parameter | UV-Vis Spectroscopy | HPLC-DAD | 1H qNMR |
|---|---|---|---|
| Analysis Time | 10-15 minutes per sample | 30-40 minutes per sample | Significantly shorter than HPLC [81] |
| Equipment Cost | Low to moderate | High | Very high |
| Sample Preparation | Moderate (extraction required) | Extensive (extraction, filtration) | Minimal |
| Specificity | Moderate (interference possible) | High (separation before detection) | High (characteristic chemical shifts) |
| Accuracy | Good for simple matrices | Excellent | Comparable to HPLC [81] |
| Limitations | Limited to transparent solutions; matrix interference in emulsions [82] | Method development complex; longer analysis | Specialized expertise required; cost |
| Ideal Use Case | Routine quality control; rapid screening | Regulatory testing; complex matrices | When available; rapid quality control [81] |
Case Study Results from Commercial Products
Analysis of six commercial cosmetic formulations demonstrates the practical application of this methodology:
- Sample 1: Declared 1.0% bakuchiol; measured 0.51% by both UV-Vis and HPLC
- Sample 2: Declared bakuchiol content; none detected by UV-Vis or HPLC
- Sample 3: Declared 1.0% bakuchiol; measured 1.0% by both methods
- Sample 4: Highest bakuchiol content at 3.6% by both methods
- Samples 5-6: Emulsion formulations showed presence of bakuchiol but quantitative analysis challenging due to incomplete extraction [82]
These results highlight UV-Vis spectroscopy's reliability for quantifying bakuchiol in simple, oil-based formulations, with strong correlation to HPLC findings. However, emulsion-based products presented extraction challenges that limited accurate quantification, emphasizing the importance of sample preparation in method development.
Troubleshooting and Method Optimization
Common Issues and Solutions
Poor Linearity in Calibration (R² < 0.990)
- Potential cause: Improper standard preparation or instrument drift
- Solution: Verify pipette calibration, prepare fresh standards, ensure instrument proper warming time [14]
Sample Precipitation or Turbidity
- Potential cause: Incompatibility between formulation components and ethanol
- Solution: Additional centrifugation, filtration through 0.45μm membrane, or alternative solvent systems [82]
Absorbance Values Exceeding Linear Range
- Potential cause: Sample concentration too high for method range
- Solution: Appropriate dilution with ethanol to bring within calibration range [13]
Matrix Interference
- Potential cause: Other formulation components absorbing at 262 nm
- Solution: Verify specificity through standard addition method or employ secondary technique (HPLC) for confirmation [82]
Method Validation Parameters
For regulatory compliance, include these validation parameters:
- Linearity: R² ≥ 0.990 across working range
- Precision: %RSD ≤ 5% for replicate measurements
- Accuracy: 85-115% recovery in spike recovery experiments
- Limit of Quantification (LOQ): Signal-to-noise ratio ≥ 10:1
This case study demonstrates that UV-Vis spectroscopy provides a reliable, cost-effective method for quantifying bakuchiol in cosmetic formulations, particularly suitable for routine quality control applications. The methodology shows excellent agreement with HPLC for oil-based formulations but faces limitations with emulsion-based systems where extraction efficiency becomes challenging.
The calibration curve approach detailed herein offers researchers a robust framework for quantifying active compounds in complex matrices, with appropriate modifications for specific formulation challenges. Future method development should focus on improving extraction efficiencies for emulsion systems and expanding applications to other cosmetic active ingredients requiring quality control and standardization.
The pervasive contamination of ecosystems by plastic waste represents one of the most pressing environmental challenges of our time. While microplastics (1 µm to 5 mm) have been extensively documented, their smaller counterparts—nanoplastics (NPs), typically defined as plastic particles with at least one dimension below 1 µm—present a more complex analytical and toxicological problem [32]. Their minute size, high surface area-to-volume ratio, and ability to penetrate biological barriers significantly increase their potential environmental mobility and bioavailability [32] [84]. Consequently, developing robust, accessible methodologies for their detection and quantification is paramount for accurate environmental risk assessment.
A critical hurdle in nanoplastic research has been the reliance on pristine, commercially available polystyrene nanobeads, which are uniform in size, shape, and composition. These synthetic analogs share poor analogies with the highly heterogeneous, irregularly shaped, and chemically complex nanoplastics formed in nature through the top-down environmental degradation of plastic waste [84]. This disparity creates a significant gap between laboratory studies and real-world conditions, potentially compromising the relevance of toxicological and environmental fate data.
This case study details the application of UV-Visible (UV-Vis) spectroscopy for the quantification of environmentally relevant, true-to-life nanoplastics (T2LNPs). We focus on a protocol for generating T2LNPs from common plastic products and systematically evaluate UV-Vis spectroscopy against established mass- and number-based quantification techniques. The work is contextualized within a broader thesis on developing UV-Vis calibration curves for compound quantification, demonstrating its viability as a rapid, accessible, and non-destructive tool for environmental nanoplastic research [32].
True-to-Life Nanoplastics: Generation and Significance
The Case for True-to-Life Models
The limitations of synthetic nanobeads extend beyond their physical homogeneity. Studies have shown that the biological identity of a nanoparticle, imparted by the layer of biomolecules (the "protein corona") that adsorbs to its surface upon entering a biological fluid, dictates its subsequent interactions with cells and tissues. Research has demonstrated that T2LNPs, with their irregular surfaces and complex chemistries, adsorb a distinctly different protein corona from human plasma compared to synthetic nanobeads [84]. Since the corona is what a "cell sees," this finding suggests that T2LNPs are essential for obtaining biologically relevant data on nanoplastic impacts [84].
Protocol: Generation of True-to-Life Polystyrene Nanoplastics
The following protocol, adapted from Ducoli et al. (2022 & 2025), describes the production of T2LNPs from polystyrene disposable items via mechanical fragmentation [32] [84].
Principle: Plastic items are embrittled and physically fragmented under cryogenic conditions to mimic the mechanical weathering processes that generate nanoplastics in the environment, resulting in heterogeneous, polydisperse particles.
Materials and Equipment:
- Source Plastic: White, unpigmented polystyrene disposable items (e.g., coffee cups).
- Size Reduction: Ultracentrifugal mill (e.g., Retsch ZM 200) equipped with a 120 μm mesh sieve.
- Cryogen: Liquid nitrogen.
- Solvent: Milli-Q water, pre-treated by high-speed centrifugation (16,000 × g for 45 min) to remove particulate background interference.
- Cleaning Supplies: Laboratory detergent, sonication water bath.
- Centrifuges: Microcentrifuge and high-speed centrifuge (capable of ≥16,000 × g)
Procedure:
- Preparation: Manually cut the plastic items into small pieces (~1 cm²). Wash them thoroughly with Milli-Q water and detergent, followed by external sonication for 15 minutes. Perform three rinsing cycles with Milli-Q water and air-dry.
- Cryo-Embrittlement: Submerge the plastic pieces in liquid nitrogen for at least 30 minutes.
- Mechanical Fragmentation: Transfer the frozen pieces to an ultracentrifugal mill pre-cooled with liquid nitrogen. Fragment the plastic at 18,000 rpm for approximately 60 seconds under a continuous flow of liquid nitrogen.
- Collection: Collect the resulting plastic powder, which has passed through the 120 μm sieve.
- Nanoplastic Separation: Suspend ~0.4 g of the powder in 30-50 mL of pre-treated Milli-Q water and externally sonicate for 30 minutes to disperse the nanoplastics.
- Sequential Centrifugation:
- Step 1: Centrifuge the suspension at 500 × g for 45 minutes. This pellets large microplastics.
- Step 2: Carefully collect the supernatant and centrifuge it at 16,000 × g for 45 minutes. This pellets the T2LNPs.
- Resuspension: Discard the final supernatant and resuspend the T2LNP pellet in a known volume of pre-treated Milli-Q water to create a concentrated stock suspension.
Quantitative Analysis Using UV-Visible Spectroscopy
Theoretical Basis
UV-Vis spectroscopy measures the absorption of light by a compound in solution or suspension. The fundamental principle is the Beer-Lambert Law [18]: [ A = \varepsilon b c ] where:
- ( A ) is the measured absorbance (unitless),
- ( \varepsilon ) is the molar absorptivity (M⁻¹cm⁻¹),
- ( b ) is the path length of the cuvette (cm),
- ( c ) is the concentration of the analyte (M).
For nanoplastics, which are complex suspensions of particles rather than molecular solutions, the absorbance signal is a composite of both genuine light absorption by the polymer and light scattering by the particles. Nevertheless, for a given set of particle characteristics (size, shape, polymer type), the measured absorbance can be empirically correlated with concentration, making UV-Vis a practical quantification tool [32].
Protocol: Creating a UV-Vis Calibration Curve and Quantifying Unknowns
This protocol outlines the steps for quantifying nanoplastics using a microvolume UV-Vis spectrophotometer, which is ideal for scarce samples [32] [14].
Materials and Equipment:
- UV-Vis spectrophotometer (single/double beam or diode array).
- Quartz or UV-transparent plastic microvolume cuvettes.
- Precision pipettes and tips.
- Volumetric flasks or microtubes.
- Standard solution of known concentration (e.g., fragmented T2LNP stock or commercial nanobeads).
- Solvent (pre-treated Milli-Q water).
- Computer with data analysis software (e.g., Excel, Origin).
Procedure:
- Prepare Stock Standard: Use a well-characterized T2LNP stock suspension or a commercial nanobead suspension of known concentration as the primary standard.
- Prepare Calibration Standards: Perform a serial dilution to create a series of at least five standard solutions spanning the expected concentration range of the unknown samples.
- Pipette a specific volume of the stock solution into the first volumetric flask/microtube.
- Add solvent to the mark and mix thoroughly. This is Standard 1.
- Pipette a volume from Standard 1 into the next vessel, add solvent, and mix to create Standard 2.
- Repeat this process to create a dilution series. A minimum of five standards is recommended for a reliable curve [14].
- Measure Blank and Standards:
- Place a cuvette filled with the solvent (pre-treated Milli-Q water) into the spectrophotometer and record a baseline or "blank" measurement to zero the instrument.
- For each standard, pipette a microvolume (typically 1-2 µL) into the sample pedestal or load it into a cuvette. Measure the absorbance across a relevant wavelength range (e.g., 200-800 nm).
- Identify the wavelength of maximum absorbance (( \lambda{max} )) from the full spectrum. Record the absorbance at this ( \lambda{max} ) for all standards. Obtain three to five replicate readings per standard for statistical rigor [14].
- Plot the Calibration Curve:
- In a spreadsheet, plot the average absorbance (y-axis) against the corresponding known concentration (x-axis) for each standard.
- Perform a linear regression analysis to fit the data to the equation ( y = mx + b ), where ( m ) is the slope and ( b ) is the y-intercept.
- Calculate the coefficient of determination (R²). A value of 0.9 or better is typically considered acceptable, indicating a strong linear relationship [18] [14].
- Analyze Unknown Samples:
- Prepare the unknown environmental or test samples in the same solvent matrix as the standards.
- Measure the absorbance of the unknown at the same ( \lambda_{max} ).
- Use the linear equation from the calibration curve to calculate the concentration of the unknown: ( c{unknown} = (A{unknown} - b) / m ).
Workflow and Analytical Context
The following diagram illustrates the integrated workflow for T2LNP analysis, positioning the UV-Vis quantification protocol within a broader, multi-technique analytical strategy.
Comparative Analytical Techniques for Validation
To assess the performance of UV-Vis spectroscopy, its results must be benchmarked against established orthogonal methods. The following table summarizes key techniques used for the characterization and quantification of nanoplastics.
Table 1: Orthogonal Techniques for Nanoplastic Characterization and Quantification
| Technique | Measurement Principle | Key Outputs | Advantages | Limitations |
|---|---|---|---|---|
| Pyrolysis GC-MS (Py-GC-MS) [32] [85] | Thermal decomposition & mass spectrometry | Polymer-specific mass concentration | High chemical specificity; identifies polymer type. | Destructive; no size/shape information; complex operation. |
| Thermogravimetric Analysis (TGA) [32] | Mass loss upon heating | Mass concentration | Overcomes size limitations; provides mass data. | Destructive; no information on size, shape, or color. |
| Nanoparticle Tracking Analysis (NTA) [32] [85] | Light scattering & Brownian motion | Particle size distribution & number concentration | Provides number-based data; visualizes particles. | Limited effectiveness with highly polydisperse/irregular samples. |
| Asymmetrical Flow Field-Flow Fractionation (AF4) [86] [85] | Size-based separation in a channel | Size-resolved separation of particles | Excellent for polydisperse samples; can be coupled to multiple detectors (e.g., MALS, UV). | High instrumentation cost; complex method development. |
| Raman Microspectroscopy [87] [85] | Inelastic light scattering | Chemical identification & particle imaging | High molecular specificity; non-destructive; can identify pigment types. | Can be affected by fluorescence; complex data analysis. |
Key Findings from Comparative Studies
A 2025 comparative analysis demonstrated that UV-Vis spectroscopy provides a rapid, accessible, and effective means of quantifying true-to-life polystyrene nanoplastics in stock suspensions [32]. While the study noted some underestimation of concentration compared to mass-based techniques like Py-GC/MS and TGA, the UV-Vis results were consistent in terms of order of magnitude and showed reliable trends across different samples [32]. This confirms its utility as a valuable screening and quantification tool, especially when sample volumes are limited, and sample conservation for subsequent analysis is critical.
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of T2LNP research requires specific reagents and equipment. The following table details the essential components of the research toolkit.
Table 2: Essential Research Reagents and Materials for T2LNP Analysis
| Item | Function/Application | Key Considerations |
|---|---|---|
| Ultracentrifugal Mill | High-energy size reduction of source plastics under cryogenic conditions. | Cryogenic operation is essential to prevent polymer degradation and generate true-to-life particles [32] [84]. |
| Liquid Nitrogen | Cryogen for embrittling plastic prior to milling. | Ensures brittle fracture, mimicking environmental mechanical weathering. |
| Pre-treated Milli-Q Water | Solvent for nanoplastic suspensions and dilutions. | Must be pre-centrifuged (e.g., 16,000 × g) to remove background particulate interference [84]. |
| Surfactant (e.g., NovaChem 100) | Additive to standard solutions to prevent nanoplastic aggregation. | Critical for maintaining particle stability during analysis by techniques like AF4 [86]. |
| Microvolume UV-Vis Spectrophotometer | Quantification of nanoplastic concentrations via absorbance. | Ideal for scarce samples; allows for sample recovery [32]. |
| Quartz Cuvettes | Sample holders for UV-Vis spectroscopy. | Required for measurements in the UV range; compatible with a wide wavelength spectrum. |
| Precision Pipettes & Volumetric Flasks | Accurate preparation of standard solutions and sample dilutions. | Essential for creating an accurate and reliable calibration curve [14]. |
| Polystyrene Reference Materials | Commercial nanospheres used as a baseline model or for calibration. | Useful for method development but lack the environmental relevance of T2LNPs [32] [84]. |
This case study establishes a validated protocol for the generation and quantification of true-to-life nanoplastics, emphasizing the practical application of UV-Vis spectroscopy. The comparative data confirms that while traditional, high-end techniques remain crucial for definitive characterization and validation, UV-Vis spectroscopy holds significant value as a rapid, accessible, and non-destructive primary quantification method. By employing T2LNPs, researchers can bridge the gap between simplified laboratory models and the complex reality of environmental plastic pollution, thereby generating more ecologically relevant data for risk assessment and regulatory decision-making.
The accurate quantification of chemical compounds is a cornerstone of research and development in pharmaceuticals, forensics, and environmental science. Among the plethora of available analytical techniques, Ultraviolet-Visible (UV-Vis) spectroscopy, High-Performance Liquid Chromatography (HPLC), and Nuclear Magnetic Resonance (NMR) spectroscopy are widely employed, each with distinct advantages and limitations. This article provides a comparative analysis of these three techniques, framed within the context of developing robust UV-Vis calibration curves for compound quantification. The critical need for reliable quantification methods is underscored by applications ranging from determining drug purity in forensic analysis [88] to quantifying active ingredients in cosmetics [82] and environmental pollutants [32]. Understanding the complementary strengths of these methods allows scientists to select the optimal tool or combination of tools for their specific analytical challenges.
Theoretical Principles and Quantitative Foundations
The fundamental principles governing UV-Vis, HPLC, and NMR differ significantly, directly impacting their application in quantitative analysis.
UV-Vis Spectroscopy relies on the measurement of the absorption of ultraviolet or visible light by a molecule. When a molecule absorbs light of a specific wavelength, electrons are promoted from a ground state to an excited state. The Beer-Lambert Law (A = εlc) forms the quantitative foundation, establishing a linear relationship between absorbance (A) and the concentration (c) of the analyte, provided the path length (l) and molar absorptivity (ε) are constant. The requirement for a chromophore—a functional group that absorbs UV or Vis light—is a primary limitation, as molecules lacking these groups cannot be detected directly [82] [89].
HPLC is a separation technique that distinguishes compounds based on their differential partitioning between a mobile phase (liquid) and a stationary phase (packed inside a column). Quantification is achieved by coupling the chromatographic separation with a detector, most commonly a UV-Vis spectrophotometer. The area or height of a chromatographic peak is proportional to the concentration of the analyte. HPLC's power lies in its ability to physically separate and individually quantify multiple components in a complex mixture, a task where direct UV-Vis spectroscopy often fails [82] [89].
NMR Spectroscopy exploits the magnetic properties of certain atomic nuclei (e.g., ^1H, ^13C). When placed in a strong magnetic field, these nuclei can absorb radiofrequency radiation. The resulting NMR spectrum provides detailed information on the chemical environment, connectivity, and dynamics of every atom in the molecule. For quantification, the integral of an NMR signal is directly proportional to the number of nuclei giving rise to that signal. This inherent quantifiability allows for absolute concentration determination without the need for a calibration curve, using an internal standard of known concentration [88] [82]. Benchtop NMR spectrometers, now more accessible, maintain this quantitative capability, albeit with lower sensitivity and resolution than high-field instruments [88].
Comparative Technical Performance
The selection of an analytical method depends on a balanced assessment of its performance characteristics against the requirements of the analysis. The table below summarizes the key parameters for UV-Vis, HPLC, and NMR.
Table 1: Comparative Analysis of Key Performance Parameters for UV-Vis, HPLC, and NMR
| Parameter | UV-Vis Spectroscopy | HPLC (with UV detection) | NMR Spectroscopy |
|---|---|---|---|
| Quantitative Principle | Beer-Lambert Law | Calibration Curve (Peak Area) | Signal Integration (vs. Internal Standard) |
| Sensitivity | Good for chromophores [89] | Superior; can detect low-level impurities [89] | Relatively low; requires microgram to milligram amounts [90] |
| Selectivity/Specificity | Limited; prone to interferences [89] | High; excellent separation capabilities [89] | Unparalleled; provides atom-specific information [90] |
| Structural Information | Limited to chromophore presence | Limited (retention time only) | Comprehensive (connectivity, stereochemistry) [90] |
| Analysis of Mixtures | Difficult without separation | Excellent for complex mixtures [82] | Possible with advanced processing (e.g., QMM) [88] |
| Analysis Speed | Fast (seconds to minutes) [89] | Moderate (minutes to hours) [89] | Minutes for simple 1D, to hours/days for complex 2D [90] |
| Sample Preparation | Minimal [89] | Often extensive (extraction, filtration) [90] | Minimal (dissolution in deuterated solvent) [90] |
| Cost & Accessibility | Low cost; simple setup [89] | High cost; complex instrumentation [89] | Very high cost; requires specialized maintenance [90] |
The quantitative performance of these techniques has been directly compared in recent research. A 2025 study on benchtop NMR for methamphetamine analysis used a Quantum Mechanical Model (QMM) to achieve a Root Mean Square Error (RMSE) of 1.3 mg/100 mg, a performance comparable to HPLC-UV which had an RMSE of 1.1 mg/100 mg [88]. Another 2025 study on bakuchiol quantification in cosmetics found that ¹H qNMR provided results comparable to HPLC analysis, with the added benefit of a significantly shorter analysis time [82]. UV-Vis, while rapid and accessible, can be unreliable for complex samples; in the same bakuchiol study, it failed to quantify two emulsion-based samples due to incomplete dissolution and potential interference [82].
Application Notes & Experimental Protocols
Protocol 1: Developing a UV-Vis Calibration Curve for a Pure Compound
This protocol outlines the steps for creating a calibration curve for a chromophore-containing compound like bakuchiol [82] or a drug substance in a simple matrix.
1. Reagent and Solution Preparation:
- Standard Stock Solution: Accurately weigh approximately 10 mg of the pure analyte reference standard. Transfer to a volumetric flask and dissolve, making up to volume with an appropriate solvent (e.g., ethanol, methanol) to obtain a known stock concentration (e.g., 1 mg/mL).
- Standard Working Solutions: Perform a serial dilution of the stock solution to prepare at least five standard solutions covering the expected concentration range of the sample (e.g., 2, 4, 6, 8, 10 µg/mL).
2. Instrumental Procedure:
- Zero the UV-Vis spectrophotometer using a cuvette filled only with the solvent (blank).
- Scan the standard solutions across a relevant wavelength range (e.g., 200-400 nm) to identify the wavelength of maximum absorption (λ_max).
- At the determined λ_max, measure the absorbance of each standard working solution in triplicate.
3. Calibration and Quantification:
- Calculate the average absorbance for each concentration.
- Plot the average absorbance (y-axis) against the corresponding concentration (x-axis) and perform linear regression analysis. The resulting equation (y = mx + c) is your calibration curve.
- Measure the absorbance of your unknown sample solution at the same λ_max and use the calibration curve equation to calculate its concentration.
Protocol 2: HPLC-UV Method for Quantifying Compounds in a Complex Mixture
This method is essential when the target analyte is part of a formulation with multiple ingredients, such as in cosmetic serums or pharmaceutical tablets [82].
1. Chromatographic Conditions:
- Column: Reversed-phase C18 column (e.g., 250 mm x 4.6 mm, 5 µm).
- Mobile Phase: Acetonitrile and water, often modified with an acid like formic acid (1%). Use isocratic or gradient elution based on complexity.
- Flow Rate: 1.0 mL/min.
- Detection: UV-Vis Diode Array Detector (DAD), wavelength set to the analyte's λ_max (e.g., 260-262 nm for bakuchiol).
- Injection Volume: 10-20 µL.
2. Sample and Standard Preparation:
- Standard Solution: Prepare a series of standard solutions of the pure analyte at known concentrations.
- Sample Preparation: For a cosmetic serum or a crushed tablet, accurately weigh a portion of the sample. Extract the analyte using an appropriate solvent (e.g., ethanol, acetonitrile) via sonication and vortex mixing. Centrifuge and filter the supernatant through a 0.45 µm membrane filter before injection.
3. System Suitability and Quantification:
- Prior to sample analysis, inject the standard solutions to ensure the system is suitable (e.g., peak symmetry, resolution, and repeatability meet predefined criteria).
- Inject the prepared sample solutions. Identify the analyte based on its retention time compared to the standard.
- Quantify the analyte in the sample by comparing the peak area (or height) to the calibration curve generated from the standard solutions.
Protocol 3: Quantitative ¹H NMR (qNMR) for Absolute Quantification
qNMR is a powerful primary method that can be used to quantify compounds, including those without a strong chromophore, and does not require identical reference standards [88] [82].
1. Sample Preparation:
- Select a suitable internal standard (IS) with a simple, non-overlapping signal (e.g., nicotinamide, maleic acid). Accurately weigh the IS and the sample into the same NMR tube.
- Dissolve the mixture in a deuterated solvent (e.g., CDCl₃, DMSO-d₆) to a known total volume. Ensure the solution is homogeneous.
2. NMR Data Acquisition:
- Using a benchtop (e.g., 60 MHz) or high-field NMR spectrometer, acquire a ¹H NMR spectrum with a sufficient number of scans to ensure a good signal-to-noise ratio.
- Use a long relaxation delay (d1 > 5 * T1 of the slowest relaxing nucleus) to ensure complete longitudinal relaxation and quantitatively accurate integrals.
3. Data Processing and Calculation:
- Process the Free Induction Decay (FID) with exponential line broadening and phase correction.
- Precisely integrate the selected signal from the analyte and the chosen signal from the internal standard.
- Calculate the purity or concentration of the analyte using the formula:
[Analyte] = (I_A / I_IS) * (N_IS / N_A) * (MW_A / MW_IS) * (W_IS / W_A) * P_ISWhere I = Integral, N = Number of protons, MW = Molecular Weight, W = Weight, P = Purity, and subscripts A and IS refer to analyte and internal standard, respectively. For complex mixtures, advanced processing like Quantitative Global Spectral Deconvolution (qGSD) or Quantum Mechanical Modelling (QMM) can be applied to deconvolute overlapping signals [88].
Workflow Visualization
The following diagram illustrates a strategic workflow for selecting and applying UV-Vis, HPLC, and NMR in quantitative analysis, highlighting their complementary roles from rapid screening to definitive confirmation.
Diagram 1: Strategic Workflow for Quantitative Technique Selection
Research Reagent Solutions
The following table lists essential materials and reagents required for the quantitative analyses described in the protocols.
Table 2: Essential Research Reagents and Materials for Quantitative Analysis
| Item | Function / Application | Example / Specification |
|---|---|---|
| Analytical Reference Standard | Serves as the known substance for calibration curve generation in UV-Vis and HPLC, and for method validation. | High-purity compound (e.g., >98% bakuchiol, methamphetamine HCl) [88] [82]. |
| Internal Standard (for qNMR) | Provides a reference signal with known proton count and concentration for absolute quantification in NMR. | Nicotinamide, maleic acid, or other compounds with a simple, non-overlapping signal [82]. |
| Deuterated Solvents | Allows for NMR frequency lock and prevents large solvent signals from dominating the ¹H spectrum. | CDCl₃, DMSO-d₆ [82]. |
| HPLC-Grade Solvents | Used as the mobile phase and for sample preparation. High purity is critical to prevent baseline noise and column damage. | Acetonitrile, Methanol, Water (with 0.1% Formic Acid) [82] [89]. |
| Chromatography Column | The stationary phase for HPLC where the separation of mixture components occurs. | Reversed-phase C18 column (e.g., 250 mm x 4.6 mm, 5 µm) [82]. |
| Syringe Filters | For clarifying and purifying sample solutions prior to HPLC injection by removing particulate matter. | 0.45 µm or 0.22 µm pore size, nylon or PTFE membrane [89]. |
UV-Vis spectroscopy, HPLC, and NMR spectroscopy form a powerful, complementary toolkit for quantitative chemical analysis. UV-Vis remains the most accessible and rapid technique for routine quantification of chromophore-containing compounds in simple matrices. HPLC-UV excels at resolving and quantifying individual components in complex mixtures, making it indispensable for impurity profiling and formulation analysis. NMR spectroscopy provides the highest level of structural confirmation and enables absolute quantification without identical reference standards, making it a powerful orthogonal method.
The choice of technique is not mutually exclusive. A robust analytical strategy often involves using HPLC for separation and purity analysis, followed by NMR for definitive structural confirmation of isolated compounds or for quantifying mixtures with overlapping UV signals through advanced processing like QMM [88]. As benchtop NMR technology continues to advance, its integration into routine quantitative workflows alongside UV-Vis and HPLC will undoubtedly expand, offering scientists a more versatile and comprehensive arsenal for tackling diverse analytical challenges.
In the realm of analytical chemistry and pharmaceutical development, the quantification of specific compounds within complex matrices is a cornerstone of research and quality control. UV-Visible (UV-Vis) spectrophotometry remains a widely employed technique for such analyses due to its simplicity, cost-effectiveness, and robustness. Instrument validation is essential for determining the condition of your instrument and ensuring that products meet expected quality standards [17]. The reliability of data generated using this technique, however, is entirely contingent upon a rigorous assessment of the analytical method's performance, specifically its accuracy, precision, and specificity. These validation parameters, when properly established within the framework of a calibration curve, provide scientists and drug development professionals with the confidence to make critical decisions based on their analytical results. This document outlines detailed protocols and application notes for evaluating these fundamental performance characteristics, framed within the context of developing UV-Vis calibration curves for compound quantification.
Theoretical Foundations
The Role of the Calibration Curve
A calibration curve, also known as a standard curve, is a fundamental tool used to identify the concentration of an unknown substance in a sample. It is generated by measuring the instrumental response (e.g., absorbance) from a series of standard solutions at known concentrations and fitting this data to a predictive model [13]. According to the Beer-Lambert Law, there is a linear relationship between the absorbance (A) of a sample and its concentration (c), expressed as A = ε × c × l, where ε is the molar absorptivity and l is the path length [91] [13]. This linear relationship is the foundation for quantitative analysis. The primary goal of calibration is to ensure good analytical findings and quality assurance [13].
Key Performance Parameters
For any analytical method to be deemed fit-for-purpose, it must be validated against a set of predefined criteria. The International Conference on Harmonisation (ICH) guidelines provide a framework for this validation. Among the most critical parameters are:
- Accuracy: This expresses the closeness of agreement between the measured value and a value accepted as a true or reference value. It is typically reported as percent recovery of a known, spiked amount of analyte.
- Precision: This refers to the closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions. It can be further divided into repeatability (intra-day precision) and intermediate precision (inter-day precision, different analysts, different instruments) and is expressed as relative standard deviation (RSD).
- Specificity: The ability of the method to assess the analyte unequivocally in the presence of other components, such as impurities, degradants, or matrix components, that may be expected to be present.
The following workflow illustrates the logical relationship between the calibration curve and the assessment of these key method performance parameters:
Experimental Protocols
Reagent Solutions and Materials
The following table details essential reagents and materials required for the development and validation of a UV-Vis spectrophotometric method.
Table 1: Key Research Reagent Solutions and Materials
| Item | Function / Purpose | Example / Specification |
|---|---|---|
| Standard Solution | A pure, known chemical substance used to prepare the calibration curve. | High-purity analyte (e.g., % purity 98.80-99.92) [91]. |
| Solvent for Dilution | To dissolve and dilute the standard and sample; should not absorb in the selected wavelength range. | Methanol, distilled water, or buffer solutions [91]. |
| Pipettes and Tips | For accurate and precise volumetric transfer of solutions. | Variable volume micropipettes covering required volume range. |
| Volumetric Flasks | For precise preparation of standard and sample solutions. | Class A glassware (e.g., 10, 25, 50, 100 mL). |
| UV-Vis Spectrophotometer | Instrument to measure the absorbance of solutions at specific wavelengths. | Double-beam instrument with matched quartz cells [91]. |
| Validation Filters/Lamps | Tools for instrument validation (wavelength accuracy, stray light). | Deuterium lamp (emission lines at 486.0 nm, 656.1 nm), sodium iodide solution [17]. |
Step-by-Step Workflow for Method Validation
The comprehensive workflow for developing and validating a UV-Vis spectrophotometric method, from instrument preparation to final quantification, is outlined below.
Protocol 1: Instrument Validation
Before analytical method validation, ensure the spectrophotometer itself is performing adequately [17].
- Wavelength Accuracy: Use the emission lines of a deuterium lamp (e.g., at 656.1 nm and 486.0 nm). Measure the energy spectrum and investigate the peak wavelength. The error from the true value (e.g., detected peak at 656.2 nm gives an error of 0.1 nm) is the wavelength accuracy [17].
- Stray Light: This is light outside the specified wavelength that reaches the sample. Use an aqueous solution of sodium iodide (NaI), which does not transmit light at 220 nm. Measure the transmittance first with a shutter block (X) and then with the NaI solution (Y). The stray light is defined as (Y - X). High stray light causes significant errors at high absorbances [17].
Protocol 2: Preparation of Standard Stock and Calibration Solutions
- Standard Stock Solution: Accurately weigh a sufficient quantity of the high-purity reference standard (e.g., 100 mg). Transfer it to a 100 mL volumetric flask, dissolve, and dilute to volume with an appropriate solvent (e.g., methanol) to obtain a stock solution of, for example, 1000 µg/mL [91].
- Calibration Standards: Perform serial dilutions of the stock solution to prepare at least five standard solutions covering the expected concentration range of the analyte. For instance, for an analyte with a working range of 4-20 µg/mL, prepare standards at 4, 8, 12, 16, and 20 µg/mL [91].
Protocol 3: Assessment of Accuracy
Accuracy is determined by recovery experiments, typically by spiking a pre-analyzed sample matrix with known quantities of the analyte.
- Prepare a sample of the matrix (e.g., placebo of a tablet formulation or tissue homogenate) at the target concentration level.
- Spike the matrix with known amounts of the standard analyte at three different levels (e.g., 50%, 100%, and 150% of the target concentration). For each level, perform a minimum of three determinations [91].
- Analyze these samples using the developed method.
- Calculate the percent recovery for each level using the formula:
- % Recovery = (Measured Concentration / Theoretical Concentration) × 100
Protocol 4: Assessment of Precision
Precision is evaluated at two levels: repeatability and intermediate precision.
- Repeatability (Intra-day Precision): Prepare six independent replicate samples of the same batch at 100% of the test concentration. Analyze all six on the same day by the same analyst. Calculate the Relative Standard Deviation (RSD) of the measured concentrations [91].
- Intermediate Precision (Inter-day Precision): To evaluate the impact of random variations, prepare and analyze the tablet samples in triplicate per day for three consecutive days, or have a second analyst repeat the method. Calculate the overall RSD from all nine determinations [91].
Protocol 5: Assessment of Specificity
Specificity ensures that the measured response is due to the analyte alone.
- For Assay: Compare the chromatograms or spectra of a blank (solvent or matrix without analyte), the standard analyte, and the sample containing the analyte in its matrix (e.g., extracted drug product with excipients) [92] [91].
- In techniques like baseline manipulation spectroscopy, specificity can be achieved by using a solution of one analyte as a blank to independently resolve the signal of the second analyte in a mixture [91].
- The method should demonstrate that the blank or matrix components do not interfere with the analyte's signal at the chosen wavelength(s).
Data Analysis and Acceptance Criteria
After conducting the experiments, the collected data must be systematically evaluated against predefined acceptance criteria to determine the method's validity.
Table 2: Summary of Validation Parameters and Acceptance Criteria
| Performance Parameter | Experimental Procedure | Recommended Acceptance Criteria | Exemplary Data from Literature |
|---|---|---|---|
| Calibration Curve Linearity | Plot absorbance vs. concentration. Perform linear regression. | Correlation coefficient (r) > 0.998. Fischer variance ratio test for linearity [91]. | Correlation coefficient of 0.9999 reported for Sofosbuvir [92]. |
| Accuracy | Recovery study at 3 levels (50%, 100%, 150%) with multiple determinations. | Mean recovery between 98–102% [91]. | RSD for recovery ranging from 0.67% to 9.42% across different media [92]. |
| Precision (Repeatability) | Six replicate measurements of a single homogeneous sample. | RSD ≤ 2.0% [91]. | Coefficient of variation (RSD) below 2% achieved for tablet formulation [91]. |
| Specificity | Compare analyte response in the presence of matrix components. | No interference from blank or matrix at the analyte retention time/wavelength. | Baseline manipulation method successfully resolved Drotaverine and Etoricoxib in a mixture [91]. |
| Sensitivity (LOD & LOQ) | LOD = (3.3 × σ) / b; LOQ = (10 × σ) / b; where σ is std. dev. of response, b is slope. | Signal-to-noise ratio of 3:1 for LOD and 10:1 for LOQ. | LOD/LOQ for Sofosbuvir: 0.27 µg/mL and 0.83 µg/mL in liver tissue [92]. |
The rigorous assessment of accuracy, precision, and specificity is non-negotiable for developing a reliable and robust UV-Vis spectrophotometric method for compound quantification. By adhering to the structured experimental protocols and acceptance criteria outlined in these application notes, researchers and drug development professionals can ensure the integrity of their analytical data. A properly validated method, underpinned by a linear calibration curve and a well-characterized spectrophotometer, provides a solid foundation for critical decisions in pharmaceutical research, quality control, and beyond, ultimately guaranteeing the safety and efficacy of the final product.
Conclusion
Mastering UV-Vis calibration is fundamental for reliable quantification in drug development and biomedical research. This guide synthesizes that while the foundational Beer-Lambert Law and linear regression are powerful, modern applications benefit from exploring non-linear models and rigorous validation to extend the usable concentration range and improve accuracy. Future directions involve integrating AI for data analysis, developing standardized protocols for complex biological matrices, and further establishing UV-Vis as a rapid, complementary technique to more complex methods like HPLC-MS in the analytical toolkit. Embracing these advanced practices will significantly enhance the quality and reliability of quantitative data, accelerating research and development outcomes.
References
- 1. Beer-Lambert Law | Transmittance & Absorbance [https://www.edinst.com/resource/the-beer-lambert-law/]
- 2. UV-Vis Spectroscopy: Principle, Strengths and Limitations ... [https://www.technologynetworks.com/analysis/articles/uv-vis-spectroscopy-principle-strengths-and-limitations-and-applications-349865]
- 3. The Beer-Lambert Law [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy/Electronic_Spectroscopy_Basics/The_Beer-Lambert_Law]
- 4. Validation of the UV-Vis Spectrophotometric Method for ... [https://oahsj.org/index.php/oahsj/article/view/101]
- 5. Beer-Lambert's Law: Principles and Applications in Daily Life [https://www.findlight.net/blog/beer-lamberts-law-explained-applications/]
- 6. Understanding the Limits of the Bouguer-Beer-Lambert Law [https://www.spectroscopyonline.com/view/understanding-the-limits-of-the-bouguer-beer-lambert-law]
- 7. Development and validation of a green UV-Vis ... [https://pubmed.ncbi.nlm.nih.gov/41092591/]
- 8. Development and validation of a green UV–Vis ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814625038294]
- 9. Beer–Lambert law for optical tissue diagnostics [https://pmc.ncbi.nlm.nih.gov/articles/PMC8553265/]
- 10. UV Vis Spectrophotometer Resources [https://www.thermofisher.com/us/en/home/industrial/spectroscopy-elemental-isotope-analysis/molecular-spectroscopy/uv-vis-spectrophotometry/resources.html]
- 11. Application of Double Beam Spectrophotometer [https://www.hunterlab.com/blog/application-of-double-beam-spectrophotometer/]
- 12. Ultraviolet Detectors: Perspectives, Principles, and Practices [https://www.chromatographyonline.com/view/ultraviolet-detectors-perspectives-principles-and-practices]
- 13. What Is a Calibration Curve in a Spectrophotometer? [https://www.hunterlab.com/blog/what-is-a-calibration-curve-in-a-spectrophotometer/]
- 14. How to Make a Calibration Curve: A Step-by-Step Guide [https://www.labmanager.com/how-to-make-a-calibration-curve-28411]
- 15. Calibration Practices in Clinical Mass Spectrometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC9467832/]
- 16. 3.4: Calibration Methods [https://chem.libretexts.org/Courses/University_of_San_Diego/USD_CHEM_220%3A_Fall_2022_(Gillette)/03%3A_Basic_Analytical_Tools/3.04%3A_Calibration_Methods]
- 17. Instrument Validation and Inspection Methods [https://www.ssi.shimadzu.com/service-support/technical-support/analysis-basics/fundamentals-uv/validation.html]
- 18. 4.4: UV-Visible Spectroscopy [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/04%3A_Chemical_Speciation/4.04%3A_UV-Visible_Spectroscopy]
- 19. 14.1: What is Molar Absorptivity? [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Instrumental_Analysis_(LibreTexts)/14%3A_Applications_of_Ultraviolet_Visible_Molecular_Absorption_Spectrometry/14.01%3A_What_is_Molar_Absorptivity]
- 20. What is “molar absorptivity”? [https://www.ssi.shimadzu.com/service-support/faq/uv-vis/light-and-theory/9/index.html]
- 21. Molar absorptivity - (Organic Chemistry II) [https://fiveable.me/key-terms/organic-chemistry-ii/molar-absorptivity]
- 22. What are “long path” cuvettes and why are they used? ... [https://www.ssi.shimadzu.com/service-support/faq/uv-vis/cuvettes/4/index.html]
- 23. Variable pathlength cell [https://en.wikipedia.org/wiki/Variable_pathlength_cell]
- 24. Calibration Curves, Part IV: Choosing the Appropriate Model [https://www.chromatographyonline.com/view/calibration-curves-part-iv-choosing-appropriate-model]
- 25. Linearity of Calibration Curves for Analytical Methods [https://www.intechopen.com/chapters/58596]
- 26. Analyzing experimental UV–vis spectra of conjugated ... [https://pubs.rsc.org/en/content/articlehtml/2025/nj/d4nj05537c]
- 27. Understanding Calibration Curves for Accurate ... [https://www.chemistryjobinsight.com/2025/03/all-about-calibration-curve.html]
- 28. UV-Vis (SOP) | PDF | Ultraviolet–Visible Spectroscopy [https://www.scribd.com/document/247911017/UV-Vis-SOP]
- 29. Calibration Methods for Volumetric Flasks [https://welchlab.com/blogs/news/calibration-methods-for-volumetric-flasks?srsltid=AfmBOoomBsGoCaqqmeN8n6_m1DXWSZK2u9hYr2g6Bm3S2jVKknoHVKKY]
- 30. Step-by-Step Guide to Calibrating Volumetric Equipment [https://www.glassment.com/guide-to-calibrating-volumetric-equipment/]
- 31. Pharma-Compliant Analysis with Flexible UV-Vis [https://www.spectroscopyonline.com/view/pharma-compliant-analysis-with-flexible-uv-vis]
- 32. Quantitative evaluation of true-to-life nanoplastics using UV ... [https://pubs.rsc.org/en/content/articlehtml/2025/en/d5en00502g]
- 33. Development and validation of a UV–Vis ... [https://www.sciencedirect.com/science/article/pii/S2215016124005703]
- 34. How to Make and Dilute Aqueous Solutions [https://www.labmanager.com/how-to-make-and-dilute-aqueous-solutions-28309]
- 35. Constructing A Calibration Curve [https://lab-manager.mydigitalpublication.com/articles/constructing-a-calibration-curve]
- 36. Optical density and absorbance measurements [https://www.bmglabtech.com/en/blog/optical-density-for-absorbance-assays/]
- 37. Errors in Spectrophotometry and Calibration Procedures to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5293527/]
- 38. Spectrophotometer Measurement Errors [https://aelabgroup.com/spectrophotometer-measurement-error/]
- 39. Chromatography Theory Calibration [https://www.chromperfect.com/post/chromatography-calibration-theory-curves-and-best-practices]
- 40. Linear regression calculator [https://www.graphpad.com/quickcalcs/linear1/]
- 41. Linear Regression in Machine learning [https://www.geeksforgeeks.org/machine-learning/ml-linear-regression/]
- 42. Utilizing tables, figures, charts and graphs to enhance the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10394528/]
- 43. Evaluating the Goodness of Instrument Calibration for ... [https://www.chromatographyonline.com/view/evaluating-the-goodness-of-instrument-calibration-for-chromatography-procedures]
- 44. 2.3.6.1. Models for instrument calibration [https://www.itl.nist.gov/div898/handbook/mpc/section3/mpc361.htm]
- 45. 5.4: Linear Regression and Calibration Curves [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Analytical_Chemistry_2.1_(Harvey)/05%3A_Standardizing_Analytical_Methods/5.04%3A_Linear_Regression_and_Calibration_Curves]
- 46. Getting a second-order polynomial trend line from a set of ... [https://stats.stackexchange.com/questions/71187/getting-a-second-order-polynomial-trend-line-from-a-set-of-data]
- 47. A Rapid UV/Vis Spectrophotometric Method for the Water ... [https://www.mdpi.com/2076-3417/10/24/9072]
- 48. How to Calibrate a Spectrophotometer: A Step-by-Step Guide [https://www.hinotek.com/how-to-calibrate-a-spectrophotometer-a-step-by-step-guide-for-accurate-scientific-measurements/]
- 49. How to Calibrate a UV-Vis Spectrophotometer: A Step-by- ... [https://www.linkedin.com/posts/muhammad-ishfaq-006b29307_3-how-to-calibrate-a-uv-visible-spectrophotometer-activity-7394579273216495616-36Ry]
- 50. Establishing a robust, versatile second-order calibration ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267025005318]
- 51. Beer-Lambert Law Spectrophotometer [https://www.hinotek.com/an-in-depth-analysis-of-the-beer-lambert-law-spectrophotometer/]
- 52. What Does Your Proficiency Testing (PT) Prove? A Look at ... [https://www.spectroscopyonline.com/view/what-does-your-proficiency-testing-pt-prove-a-look-at-inorganic-analyses-proficiency-tests-and-contamination-and-error-part-i]
- 53. Synthesis of Protium serratum fruit extract stabilized silver ... [https://www.sciencedirect.com/science/article/abs/pii/S016773222500515X]
- 54. Development and Validation of UV-Visible ... [https://pharmacophorejournal.com/article/development-and-validation-of-uv-visible-spectrophotometric-method-for-the-estimation-of-curcumin-in-bulk-and-pharmaceutical-formulation]
- 55. Ultraviolet Spectrophotometric Estimation of Nebivolol ... [https://www.ijfmr.com/research-paper.php?id=54473]
- 56. Some practical considerations for linearity assessment of ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914017305647]
- 57. Determining LOD and LOQ Based on the Calibration Curve [https://www.sepscience.com/hplc-solutions-126-chromatographic-measurements-part-5-determining-lod-and-loq-based-on-the-calibration-curve-6959]
- 58. 13.3: Effect of Noise on Transmittance and Absorbance ... [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Instrumental_Analysis_(LibreTexts)/13%3A_Introduction_to_Ultraviolet_Visible_Absorption_Spectrometry/13.03%3A_Effect_of_Noise_on_Transmittance_and_Absorbance_Measurements]
- 59. The LCGC Blog: HPLC Diagnostic Skills–Noisy Baselines [https://www.chromatographyonline.com/view/lcgc-blog-hplc-diagnostic-skills-noisy-baselines]
- 60. Fluctuations in readings when using the UV-Vis Calibration ... [https://www.mtc-usa.com/kb-article/aa-01360]
- 61. Spectrophotometer Troubleshooting [https://www.hinotek.com/spectrophotometer-troubleshooting/]
- 62. Baseline Correction | Technical Note 119 [https://www.denovix.com/tn-119-baseline-correction/]
- 63. Correcting Ultraviolet-Visible Spectra for Baseline Artifacts [https://www.sciencedirect.com/science/article/pii/S0022354923003350]
- 64. How to Interpret UV-Vis Spectroscopy Results [https://www.azooptics.com/Article.aspx?ArticleID=2768]
- 65. Calibration: Effects on Accuracy and Detection Limits in ... [https://www.spectroscopyonline.com/view/calibration-effects-on-accuracy-and-detection-limits-in-atomic-spectroscopy]
- 66. Calibration Curves [https://www.inorganicventures.com/icp-guide/calibration-curves]
- 67. Tips for Preparing Calibration Curve Standards and ... [https://www.restek.com/articles/tips-for-preparing-calibration-curve-standards-and-avoiding-sources-of-error?srsltid=AfmBOorkNPKy0O4k72Hn-nHQEYtb0JPb2p0YYpF1644HE-K7MrrBDLMp]
- 68. The clean analysis process of Mn2+ for industries: a ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-024-01367-0]
- 69. Introduction of curvature index and improvement ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267024013229]
- 70. Optimization of UV Spectrophotometric Techniques for ... [https://pubmed.ncbi.nlm.nih.gov/40825392/]
- 71. Introduction of curvature index and improvement ... [https://pubmed.ncbi.nlm.nih.gov/39788674/]
- 72. Emerging Tools for Holistic Evaluation of Analytical Methods [https://www.chromatographyonline.com/view/emerging-tools-for-holistic-evaluation-of-analytical-methods]
- 73. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 74. Analytical Method Validation: Back to Basics, Part II [https://www.chromatographyonline.com/view/analytical-method-validation-back-basics-part-ii]
- 75. Method Validation Essentials, Limit of Blank ... [https://www.biopharminternational.com/view/method-validation-essentials-limit-blank-limit-detection-and-limit-quantitation]
- 76. Development and validation of the UV-spectrophotometric method for determination of terbinafine hydrochloride in bulk and in formulation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658052/]
- 77. Approaches to Assay Characterization and Validation: Part 1 [https://www.quanterix.com/approaches-to-assay-characterization-and-validation-part-1-limit-of-detection-and-lower-limit-of-quantitation/]
- 78. What is meant by the limit of detection and quantification (LOD ... [https://mpl.loesungsfabrik.de/en/english-blog/method-validation/limit-of-detection-quantifcation]
- 79. In-line UV Spectrometry Monitoring in Cleaning Validation | Pharmaceutical Technology [https://www.pharmtech.com/view/in-line-uv-spectrometry-monitoring-in-cleaning-validation]
- 80. Comparison of approaches for assessing detection and ... [https://www.nature.com/articles/s41598-024-83474-5]
- 81. Comparison of Quantification Using UV-Vis, NMR, and ... [https://pubmed.ncbi.nlm.nih.gov/40724887/]
- 82. Comparison of Quantification Using UV-Vis, NMR, and ... [https://www.mdpi.com/1422-0067/26/14/6638]
- 83. Decoding Cosmetic Complexities: A Comprehensive Guide to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10818968/]
- 84. A different protein corona cloaks “true-to-life” nanoplastics with ... [https://pubs.rsc.org/en/content/articlehtml/2022/en/d1en01016f]
- 85. Physicochemical characterization and quantification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10284950/]
- 86. Validated method for polystyrene nanoplastic separation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10328892/]
- 87. A Review of Spectroscopic Techniques used for the ... [https://www.spectroscopyonline.com/view/a-review-of-spectroscopic-techniques-used-for-the-quantification-and-classification-of-microplastics-and-nanoplastics-in-the-environment]
- 88. Comparative Analysis of Benchtop NMR and HPLC‐UV for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12500361/]
- 89. Comparative Study of UV And HPLC Methods for ... [https://www.ijsrtjournal.com/article/Comparative+Study+of+UV+And+HPLC+Methods+for+Estimation+of+Drug]
- 90. Differences in HPLC and NMR: Structural Elucidation ... [https://eureka.patsnap.com/report-differences-in-hplc-and-nmr-structural-elucidation-relevance]
- 91. Development and validation of UV-Visible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658078/]
- 92. Validation of spectrophotometric and colorimetric method ... [https://pubmed.ncbi.nlm.nih.gov/39613187/]