Electrospray Ionization Mass Spectrometry: Principles, Applications, and Optimization for Biomedical Research
This article provides a comprehensive exploration of Electrospray Ionization (ESI) for researchers and drug development professionals.
Electrospray Ionization Mass Spectrometry: Principles, Applications, and Optimization for Biomedical Research
Abstract
This article provides a comprehensive exploration of Electrospray Ionization (ESI) for researchers and drug development professionals. It covers the foundational mechanism of ESI, from charged droplet formation to gas-phase ion release. The article details its pivotal methodological applications in clinical diagnostics, drug discovery, and metabolomics, and offers systematic strategies for parameter optimization and troubleshooting. A comparative analysis with complementary ionization techniques like APPI is included to guide method selection. The review concludes by examining emerging trends and the future impact of ESI and related techniques on biomedical research and clinical applications.
The Core Mechanism of Electrospray Ionization: From Liquid Solution to Gas-Phase Ions
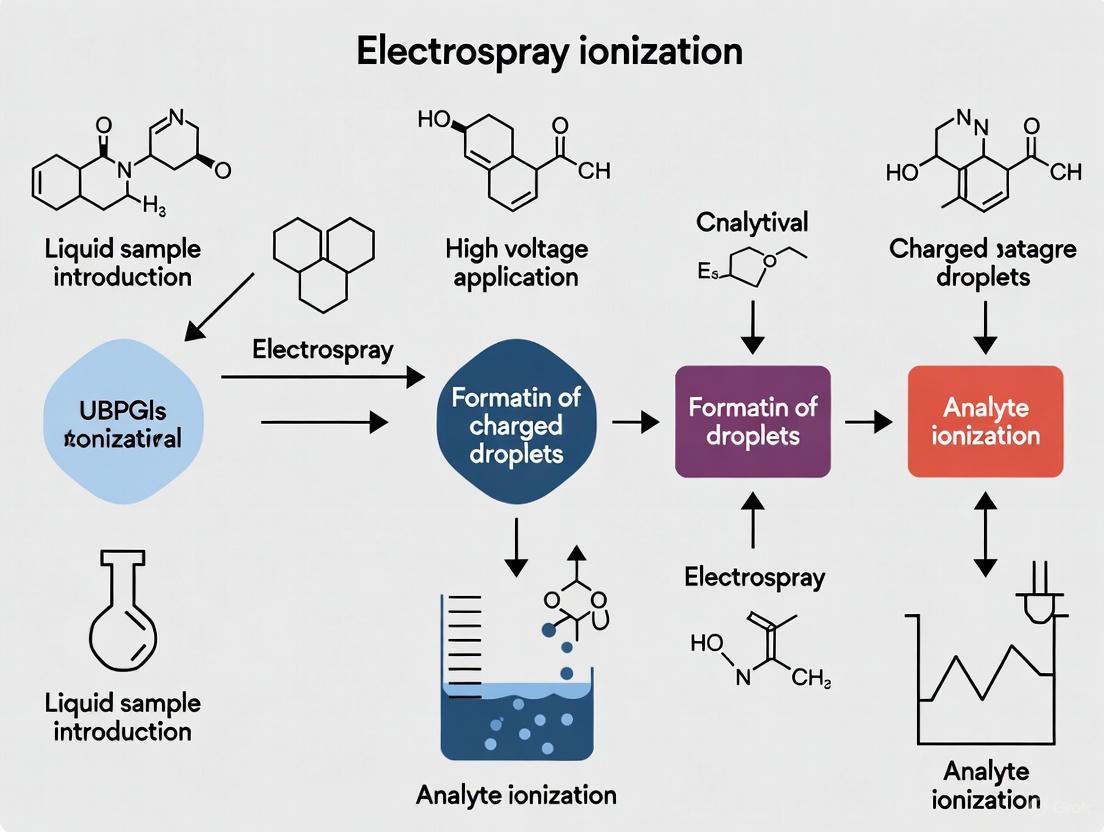
Electrospray Ionization (ESI) is a soft ionization technique that has revolutionized the analysis of biomacromolecules by enabling their transfer from a liquid solution to the gas phase as intact ions, making them amenable to mass spectrometric analysis [1]. This process is pivotal in modern research for studying proteins, peptides, and other complex biological molecules, as it overcomes their propensity to fragment and allows for the determination of very high molecular weights by producing multiply charged ions [1]. The mechanism can be fundamentally broken down into three essential steps: the dispersal of a charged aerosol, the evaporation of the solvent, and the final ejection of gas-phase ions.
The Electrospray Ionization Mechanism
The following diagram illustrates the complete ESI process, from the application of high voltage to the generation of gas-phase ions, encompassing the three core stages.
Diagram 1: The three-step ESI process, from charged droplet formation to gas-phase ion generation.
Stage 1: Droplet Formation and Dispersal
The process initiates when a sample solution is pushed through a fine metal capillary (electrospray needle) held at a high voltage, typically between 2.5 to 6.0 kV [2] [3]. This high electric field charges the surface of the liquid emerging from the tip. The mutual electrostatic repulsion between the like charges counteracts the liquid's surface tension, deforming the meniscus into what is known as a Taylor cone [1]. When the electrostatic forces overcome the surface tension, the cone's tip elongates and disperses the liquid into a fine mist or aerosol of highly charged droplets [2] [4]. The application of a nebulizing gas (such as nitrogen) shearing around the liquid stream can enhance this process, allowing for higher sample flow rates [2].
Stage 2: Solvent Evaporation and Droplet Shrinking
The cloud of charged droplets is directed towards the mass spectrometer's inlet, which is at a lower pressure. As the droplets travel, the solvent begins to evaporate, a process often assisted by a counter-current flow of a heated drying gas (like nitrogen) and the application of heat within the ESI source [1] [2]. This continuous solvent loss reduces the droplet's size while its charge remains constant. Consequently, the charge density on the droplet's surface increases significantly. This leads to an inevitable point where the electrostatic repulsion between the charges rivals the surface tension holding the droplet together—a threshold known as the Rayleigh limit [1].
Stage 3: Ion Ejection via Coulomb Fission and Desorption
Upon reaching the Rayleigh limit, the droplet becomes unstable and undergoes Coulomb fission, disintegrating into smaller, progeny droplets [1]. This "explosive" event relieves a portion of the charge (typically 10-18%) and mass (1.0-2.3%) [1]. These smaller droplets continue the cycle of solvent evaporation and Coulomb fission until the conditions are right for the final release of gas-phase ions. The mechanism for this final step is explained by two primary models [1] [3]:
- Charged Residue Model (CRM): Progeny droplets become so small that they contain, on average, one analyte molecule. The remaining solvent evaporates completely, leaving the analyte with the droplet's charges [1]. This model is generally accepted for large, folded proteins [1].
- Ion Evaporation Model (IEM): For small droplets at a critical radius, the electric field strength at the surface becomes intense enough to directly desorb (field-evaporate) solvated ions into the gas phase before the solvent fully evaporates [1]. This is thought to be the dominant mechanism for smaller ions [1].
The resulting gas-phase ions are typically protonated molecules [M+H]⁺, deprotonated molecules [M-H]⁻, or adducts with other cations like sodium [M+Na]⁺ [1]. For large macromolecules, ESI famously produces a spectrum of multiply charged ions [M+nH]ⁿ⁺, which effectively extends the mass range of the mass spectrometer and allows for the accurate mass determination of kDa-MDa molecules [1] [3].
Key Experimental Parameters and Reagent Solutions
Successful ESI analysis requires careful optimization of several parameters. The table below summarizes the core components and their functions in a typical ESI-MS setup.
Table 1: Key Research Reagent Solutions and ESI-MS Components
| Component | Function & Characteristics |
|---|---|
| Sample Solvent | Typically a mixture of water with volatile organic compounds (e.g., methanol, acetonitrile) to facilitate droplet formation and solvent evaporation [1]. |
| Acid Additives | Compounds like acetic or formic acid are added to increase solution conductivity and provide a source of protons to facilitate the ionization of analytes [1]. |
| Nebulizing Gas | An inert gas (e.g., nitrogen) that shears the liquid stream to enhance aerosol formation and stabilize the electrospray, especially at higher flow rates [2]. |
| Drying Gas | A stream of heated, inert gas (e.g., nitrogen) directed at the spray to accelerate solvent evaporation from the charged droplets [2]. |
| Metal Capillary | The emitter tip where the high voltage is applied to create the Taylor cone and generate the charged aerosol [2]. |
| High-Voltage Power Supply | Applies a high potential (2.5–6.0 kV) to the capillary relative to the spectrometer inlet, providing the electric field necessary for electrospray [2] [3]. |
Quantitative Data in ESI-MS Analysis
The unique ability of ESI to generate multiply charged ions transforms the data output, making it essential to understand the quantitative relationships for accurate mass determination.
Table 2: Quantitative Relationships in an ESI Mass Spectrum of a Protein
| Multiply Charged Ion | m/z Ratio Calculation (for Mr = 15,000 Da) | Resulting m/z |
|---|---|---|
| [M + 6H]⁶⁺ | (15000 + 6) / 6 | 2501 |
| [M + 5H]⁵⁺ | (15000 + 5) / 5 | 3001 |
| [M + 4H]⁴⁺ | (15000 + 4) / 4 | 3751 |
| [M + 3H]³⁺ | (15000 + 3) / 3 | 5001 |
| [M + 2H]²⁺ | (15000 + 2) / 2 | 7501 |
| [M + H]⁺ | (15000 + 1) / 1 | 15001 |
The molecular mass (Mr) of an unknown analyte can be calculated from two adjacent charge states in the mass spectrum, p₁ and p₂, where p₁ has the lower m/z value and a charge of z₁, and p₂ has a charge of z₁ - 1 [4]. The relevant formulas are:
- p₁ = (Mᵣ + z₁) / z₁
- p₂ = [Mᵣ + (z₁ - 1)] / (z₁ - 1)
Advanced ESI Methodologies and Protocols
Coupling with Liquid Chromatography (LC-ESI-MS)
For complex mixtures like protein digests or biological fluids, ESI is almost universally coupled with high-performance liquid chromatography (HPLC) [2]. The HPLC system acts as a front-end separation tool, fractionating the sample and introducing purified analytes directly into the ESI source. This prevents signal suppression and simplifies the mass spectrum, making it a fundamental protocol for proteomics and metabolomics [2].
Tandem Mass Spectrometry (ESI-MS/MS) for Structural Elucidation
To gain structural information, ESI is frequently paired with tandem mass spectrometry (MS/MS). A typical protocol using a triple quadrupole instrument involves [2]:
- Mass Selection: The first quadrupole (Q1) is set to isolate the precursor ion of interest.
- Collision-Induced Dissociation (CID): The selected ions are passed into a second quadrupole (Q2), which acts as a collision cell filled with an inert gas like argon. The kinetic energy of the ions is converted into vibrational energy upon collision, causing the precursor ions to fragment.
- Mass Analysis of Fragments: The resulting product (daughter) ions are mass-analyzed by the third quadrupole (Q3). This fragmentation pattern provides critical information for sequencing peptides or confirming molecular structures [2].
Common MS/MS acquisition modes include the Product Ion Scan for structural elucidation and Multiple Reaction Monitoring (MRM) for highly sensitive and specific quantitative analysis [2].
The elegant, three-step process of dispersal, evaporation, and ion ejection makes electrospray ionization a cornerstone technique in modern analytical science. Its capacity to gently bring fragile biomacromolecules into the gas phase for mass analysis has been instrumental in advancing fields from structural biology to drug discovery. By enabling the analysis of highly complex mixtures through coupling with liquid chromatography and providing deep structural insights via tandem mass spectrometry, ESI continues to be an indispensable tool in the researcher's arsenal, driving forward our understanding of biological systems at a molecular level.
Electrospray Ionization (ESI) has revolutionized the analysis of biomolecules, enabling the study of proteins, peptides, and other large, non-volatile compounds by mass spectrometry (MS) [2]. The technique transforms analytes in solution into gas-phase ions, making them amenable to mass analysis. The efficiency of this process hinges on two fundamental and interconnected electrohydrodynamic phenomena: Taylor cone formation and Coulomb fission [5]. A deep understanding of these principles is essential for researchers and drug development professionals aiming to optimize ESI-MS methods, particularly for applications in proteomics, metabolomics, and therapeutic drug monitoring[cite:3]. This guide provides a detailed examination of the theory, experimental observations, and practical methodologies surrounding these critical events.
Fundamental Theory and Definitions
The Taylor Cone
A Taylor cone is a conical deformation that forms at the surface of a conductive liquid when it is subjected to a sufficiently strong electric field [6]. This occurs at the tip of the ESI emitter, where the applied voltage creates an electric field strong enough to overcome the surface tension of the liquid.
- Formation Mechanism: When a voltage (typically 2–6 kV) is applied to a capillary tube containing the liquid sample, free charges (e.g., ions) within the liquid migrate to the liquid-air interface [6] [2]. The electric stress exerted on the surface pulls the liquid toward the counter-electrode, elongating the initially hemispherical meniscus. A stable, conical shape is achieved when the electric stress precisely balances the restoring force of the liquid's surface tension [7]. British physicist Sir Geoffrey Ingram Taylor theoretically predicted and experimentally verified this equilibrium shape in 1964, demonstrating that it is characterized by a specific semi-vertical angle of 49.3° (a whole angle of 98.6°) [6] [7].
- The Cone-Jet and Ion Emission: The stable Taylor cone represents an equilibrium state. However, when the electric field intensity at the cone's apex exceeds a critical threshold, this equilibrium is disrupted, and the cone's tip elongates into a fine liquid jet [6] [8]. It is from the apex of this jet that charged droplets are emitted into the gas phase. This "cone-jet" mode is the desired operational state for a stable electrospray [7].
Coulomb Fission (Droplet Fission)
The charged droplets produced by the Taylor cone jet undergo a process of solvent evaporation while traveling through the mass spectrometer's atmosphere-vacuum interface. As a droplet shrinks in size, its charge density increases. Coulomb fission is the process wherein a charged droplet becomes unstable and disintegrates into smaller progeny droplets.
The Rayleigh Limit: The stability limit for a charged droplet is described by Lord Rayleigh's equation [9] [5]:
z_R = 8π(ε₀γR³)¹ᐟ²Here,
z_Ris the maximum, or Rayleigh charge limit;Ris the droplet radius;γis the surface tension; andε₀is the permittivity of free space. When the charge on the droplet (z) reaches or exceeds this limit (z ≥ z_R), the Coulombic repulsion between the charges overcomes the cohesive surface tension, and the droplet undergoes fission [9].- Fission Pathways: Experimental studies, particularly using Charge Detection Mass Spectrometry (CDMS), have revealed that fission in submicrometer aqueous nanodrops is highly heterogeneous. Four distinct pathways have been identified [9]:
- Continuous Pathway: Many small progeny droplets are sequentially emitted over tens to hundreds of milliseconds.
- Prompt Pathway: A single, larger fission event ejects one or a limited number of progeny droplets carrying a significant fraction of the precursor's charge. This is the most common pathway.
- Sequential Prompt Pathway: Multiple prompt fission events occurring in sequence.
- Prefission Pathway: The emission of a few charges precedes a larger prompt fission event, analogous to "foreshocks" before an earthquake.
The continuous cycle of solvent evaporation and Coulombic fission repeatedly divides the initial droplets until the analyte ions are liberated into the gas phase, ready for mass analysis [2] [5].
Table 1: Core Principles of Taylor Cone and Coulomb Fission
| Feature | Taylor Cone | Coulomb Fission |
|---|---|---|
| Governing Physics | Balance between electric stress and surface tension | Balance between Coulombic repulsion and surface tension |
| Governing Equation | N/A (Equilibrium of forces) | Rayleigh Limit: z_R = 8π(ε₀γR³)¹ᐟ² [9] |
| Key Parameter | Semi-vertical angle of 49.3° [6] [7] | Charge-to-radius ratio (z / R) |
| Primary Outcome | Formation of a charged liquid jet and droplets [6] | Breakup of parent droplet into smaller progeny droplets [9] |
| Role in ESI | Initial droplet formation and charging | Droplet downsizing and eventual gas-phase ion release [5] |
Experimental Observations and Quantitative Data
Direct measurement of nanodrop fission became possible with techniques like Charge Detection Mass Spectrometry (CDMS). This method allows for simultaneously tracking the mass, charge, and energy per charge of individual trapped droplets over time, providing unprecedented insight into fission dynamics [9].
Key Findings from CDMS Experiments
A 2025 study using CDMS analyzed 846 trapped aqueous nanodrops, of which 154 (18.2%) underwent spontaneous fission. The charges of these droplets ranged from 44% to 158% of the Rayleigh limit, confirming that fission can occur both below and significantly above the theoretical limit [9]. The study quantified the mass and charge losses during these events, revealing the diversity of fission pathways.
Table 2: Experimental Fission Data from CDMS Studies [9]
| Fission Parameter | Observed Range | Comments |
|---|---|---|
| Frequency of Fission | 18.2% of trapped nanodrops | Charges ranged from 44-158% of the Rayleigh limit. |
| Charge Loss per Fission Event | 4% - 40% of parent charge | Varies significantly by pathway; prompt events involve larger losses. |
| Mass Loss per Fission Event | < 5% of parent mass | Generally low mass loss is a characteristic of Coulomb fission. |
| Fission Time Scale | < 1 ms to 100s of ms | Ranges from near-instantaneous (prompt) to prolonged (continuous). |
| Number of Progeny Droplets | A few to hundreds | Highly heterogeneous, dependent on the fission pathway. |
Historical and Supporting Data
Earlier theoretical and experimental work aligns with these modern observations. A 1999 model assumed an average mass loss of 2% and a charge loss of 15% per fission event based on prior optical resonance measurements [5]. This model further calculated that the size ratio of progeny droplets to the initial parent droplet is a function of the number of progeny droplets generated, highlighting the inherent variability of the process [5].
Detailed Experimental Protocols
To investigate Taylor cone formation and Coulomb fission, robust experimental setups are required. Below are detailed methodologies for key approaches.
Charge Detection Mass Spectrometry (CDMS) for Fission Analysis
This protocol is adapted from recent studies on spontaneous fission of aqueous nanodrops [9].
- Objective: To track the mass, charge, and energy per charge of individual nanodrops in real-time to observe and characterize fission events.
- Materials and Reagents:
- Nanodrop Source: Deionized water (resistivity of 18.2 MΩ·cm).
- Emitter: Borosilicate emitters (e.g., 1.0 mm outer diameter, 0.78 mm inner diameter) pulled to an inner tip diameter of ~18–20 μm.
- High Voltage Source: Capable of delivering 2.4–2.7 kV to a platinum wire inserted into the emitter.
- Instrumentation: A custom-built electrostatic ion trap charge detection mass spectrometer, equipped with:
- Heated inlet capillary (e.g., 80 °C).
- Ion funnel and sequential quadrupole ion guides.
- An electrostatic cone trap with a cylindrical detector electrode.
- A vacuum chamber housing the trap at ~1 × 10⁻⁸ Torr.
- Methodology:
- NanoESI Generation: Position the nanoESI emitter ~3 mm from the instrument inlet. Apply a voltage of 2.4–2.7 kV to the platinum wire in contact with the water to initiate electrospray and generate positively charged nanodrops [9].
- Ion Trapping: The nanodrops enter through the heated capillary, are focused by the ion funnel and quadrupoles, and are subsequently pulsed into the electrostatic cone trap. The trap is operated with a low ion current to ensure single-ion analysis and avoid ion-ion interactions.
- Data Acquisition: Trap individual nanodrops for up to 4 seconds. The current induced by the charged nanodrop on the cylindrical detector is recorded continuously. This signal is used to determine the ion's mass (
m), charge (z), and mass-to-charge ratio (m/z) throughout the trapping period. - Signal Processing: Perform Short-time Fourier Transform (STFT) analysis on the time-domain data (e.g., using a 25 ms window length and 5 ms step size) to trace oscillation frequencies, which are sensitive to changes in mass and charge.
- Data Analysis:
- Manually inspect all STFT frequency traces for sudden drops indicative of fission events.
- Classify events into fission pathways (continuous, prompt, sequential prompt, prefission).
- Calculate mass and charge losses by averaging measurements from 50 ms before and after a fission event.
- Determine the droplet diameter from the measured mass using a spherical water model.
- Compute the fraction of the Rayleigh limit from the measured charge and diameter using Equation 1.
Visualizing Taylor Cone Formation
This protocol describes a classic setup for observing the Taylor cone, based on historical and contemporary practices [6] [7].
- Objective: To generate and optically observe a stable Taylor cone in a laboratory setting.
- Materials and Reagents:
- Capillary: A metallic (e.g., stainless steel) capillary or needle with an inner diameter of 0.1 to 1 mm.
- Syringe Pump: For precise control of liquid flow rates (nL/min to μL/min).
- High Voltage DC Power Supply: Capable of delivering 2 to 20 kV.
- Counter-Electrode: A grounded metallic plate.
- Conducting Liquid: Water or an aqueous solution.
- Safety Equipment: Proper insulation, grounding, and shielding.
- Imaging System: A high-speed camera with a macro lens.
- Methodology:
- Setup: Connect the capillary to the syringe pump filled with the conducting liquid. Position the counter-electrode 5 to 50 mm away from the capillary tip, ensuring alignment.
- Initialization: Start the syringe pump at a low flow rate (e.g., 1 μL/min). Gradually increase the voltage from the power supply applied to the capillary.
- Observation: As the voltage approaches a critical threshold (e.g., 1–5 kV, depending on geometry and liquid), the meniscus will deform from hemispherical to a convex cone and finally to a stable Taylor cone with a jet emitting from its apex.
- Optimization: Adjust the flow rate and voltage to achieve a stable "cone-jet" mode. Ambient conditions like humidity should be monitored as they can affect stability.
- Validation: Capture images of the cone. Measure the semi-vertical angle to confirm it approximates the theoretical value of 49.3°.
Visualization of Processes and Workflows
The following diagrams illustrate the logical sequence of electrospray ionization and the specific experimental workflow for CDMS, as discussed in this guide.
Electrospray Ionization Process
Diagram 1: The ESI process, showing the cyclical nature of evaporation and fission leading to gas-phase ions.
CDMS Fission Analysis Workflow
Diagram 2: The workflow for analyzing droplet fission using Charge Detection Mass Spectrometry.
The Scientist's Toolkit: Essential Reagents and Materials
Successful experimentation in this field requires specific reagents and materials. The following table details key items for studying Taylor cone formation and Coulomb fission.
Table 3: Essential Research Reagents and Materials
| Item Name | Function / Role | Specifications / Examples |
|---|---|---|
| Conductive Liquid | Forms the Taylor cone and charged droplets; the medium for analyte transport. | High-purity deionized water (18.2 MΩ·cm resistivity) [9]. Solvents like methanol or acetonitrile for LC-ESI-MS. |
| ESI Emitter | The physical tip from which the Taylor cone and jet are formed. | Borosilicate glass capillaries pulled to fine tips (e.g., 18-20 μm inner diameter) [9]. Metal capillaries for some applications. |
| Supercharging Reagents | Increase the charge state of analyte ions, influencing fission dynamics and ion yield. | Sulfolane, m-nitrobenzyl alcohol (mNBA), glycerol. Typically added at <0.5% v/v [10]. |
| High-Voltage Power Supply | Provides the electric field necessary for Taylor cone formation. | Capable of delivering 2-20 kV DC [6]. |
| Syringe Pump | Precisely controls the flow rate of the liquid to the emitter for stable cone-jet operation. | Capable of delivering flow rates from nL/min to μL/min [6]. |
| Charge Detection Mass Spectrometer | The primary instrument for directly measuring mass and charge of individual droplets to study fission. | Custom-built or commercial instruments with single-ion trapping capabilities [9]. |
Generation of Multiply Charged Ions and its Impact on Analyzing Macromolecules
The analysis of macromolecules, such as proteins and protein complexes, has been revolutionized by the ability to generate multiply charged ions in the gas phase. This phenomenon effectively extends the mass range of mass spectrometers by reducing the mass-to-charge (m/z) ratios of large biomolecules, making them compatible with conventional mass analyzers [1]. Electrospray ionization (ESI) has emerged as a pivotal technique in this field, enabling the transfer of ions from solution into the gaseous phase through the application of electrical energy [2]. The multiple charging phenomenon is particularly crucial for the analysis of high-mass biological complexes, as it produces ions of relatively low m/z, making ESI amenable with mass analyzers where high m/z performance is otherwise limited [11]. The generation of multiply charged ions underpins various mass spectrometric applications, from structural characterization to quantitative measurements in clinical and pharmaceutical contexts.
The formation of multiply charged ions is not limited to ESI. Matrix-assisted laser desorption/ionization (MALDI), traditionally known for producing singly charged ions, has also been adapted under specific conditions to generate multiply charged ions. Recent research has demonstrated that homogeneous MALDI microcrystals, when prepared using methods like forced dried droplet (FDD), can produce multiply charged protein ions with charge states as high as +6 for proteins like myoglobin [12]. The control over experimental parameters such as laser fluence and matrix proton affinity (PA) plays a critical role in optimizing the charge state distributions in MALDI, with lower PA matrices (e.g., CHCA at 841 kJ/mol and Cl-CCA at 838.5 kJ/mol) generally resulting in higher charge states compared to higher PA matrices (e.g., CHCA-C3 at 879.5 kJ/mol) [12].
Mechanisms of Multiply Charged Ion Formation
Electrospray Ionization Mechanisms
The electrospray ionization process involves three fundamental steps that facilitate the transfer of ionic species from solution into the gas phase. First, a fine spray of charged droplets is dispersed from a capillary maintained at a high voltage (typically 2.5-6.0 kV). Second, solvent evaporation occurs from these charged droplets, aided by a drying gas and elevated ESI-source temperature. Third, ion ejection takes place from the highly charged droplets as they reach a critical field strength due to continuous solvent evaporation [2]. Two primary models explain the final production of gas-phase ions: the Charge Residue Model (CRM) and the Ion Evaporation Model (IEM). The CRM proposes that electrospray droplets undergo successive evaporation and fission cycles, eventually resulting in progeny droplets containing approximately one analyte ion, with gas-phase ions forming after remaining solvent molecules evaporate [1]. In contrast, the IEM suggests that when droplets reach a critical radius, the field strength at the droplet surface becomes sufficient to assist the field desorption of solvated ions [1].
For large macromolecules such as folded proteins, evidence indicates that ions form predominantly through the charged residue mechanism, while smaller ions (from small molecules) are liberated into the gas phase through the ion evaporation mechanism [1]. A third model, the Chain Ejection Model (CEM), has been proposed specifically for disordered polymers and unfolded proteins [1]. The efficiency of generating gas-phase ions varies significantly depending on compound structure, solvent composition, and instrumental parameters, with differences in ionization efficiency exceeding one million times for different small molecules [1].
Novel Charge Inversion and Ion Attachment Mechanisms
Beyond conventional ESI and MALDI, recent advances have revealed additional mechanisms for manipulating charge states. Charge inversion ion/ion reactions represent a novel approach that converts multiply charged protein cations to multiply charged protein anions via single ion/ion collisions using highly charged anions derived from nano-electrospray ionization of hyaluronic acids [11]. This process has been demonstrated with cations derived from cytochrome c, apo-myoglobin, and carbonic anhydrase, converting, for example, the [CA+22H]²²⁺ cation to a distribution of anions as high in absolute charge as [CA−19H]¹⁹⁻ [11]. This phenomenon is particularly surprising because previous studies involving reactions of multiply-charged ions of opposite polarity have shown ion/ion attachment to dominate, sometimes competing with partial proton transfer [11].
Another significant development is the multiply-charged ion attachment approach, which facilitates the mass measurement of high-mass complexes in native mass spectrometry. This method involves attaching high-mass ions of known mass and charge to populations of ions of interest, leading to well-separated signals that enable confident charge state and mass assignments from otherwise poorly resolved signals [13]. This strategy is particularly valuable for analyzing heterogeneous samples such as ribosome particles, where extensive salt adduction and inherent heterogeneity complicate mass determination [13]. The attachment of multiply-charged reagent ions to analyte complexes generates known Δm and Δz values that are much greater than those achieved through single proton or electron transfer, resulting in large m/z separations that are more readily resolved [13] [14].
Experimental Techniques and Methodologies
Electrospray Ionization Protocols
For standard ESI-MS analysis of proteins, sample preparation involves dissolving the analyte in an appropriate solvent system. A typical protocol involves preparing protein stock solutions at concentrations around 1 mg/mL in LC-MS grade water, then diluting working solutions to 5-20 μM with either pure water for near-neutral pH conditions or water with 0.5-2% acetic acid for denaturing conditions [11]. The addition of acid facilitates analyte protonation, while low surface tension solvents such as methanol promote droplet fission [15]. For nano-ESI, which operates at lower flow rates (nL/min to μL/min) and generates smaller initial droplets, improved ionization efficiency can be achieved [1]. The emitter voltage typically ranges from +1500 V for positive mode analysis of large complexes to -1400 V for negative mode analysis [13] [14].
Liquid chromatography coupling with ESI-MS requires additional optimization. When interfacing size exclusion chromatography with MS, isocratic conditions with MS-friendly mobile phases of low ionic strength are employed. For example, a protocol for separating transferrin and β-galactosidase used 50 mM ammonium acetate in LC-MS grade water as the mobile phase at a flow rate of 0.05 mL/min [16]. The column temperature is maintained at 25°C, and UV detection at 280 nm enables monitoring of the separation prior to MS analysis [16].
Ion/Ion Reaction Methodology
The experimental setup for ion/ion reactions typically employs a modified QTOF mass spectrometer capable of mutual storage of cations and anions [11] [13] [14]. The general procedure involves:
- Generating analyte ions via nano-ESI under native conditions
- Isolating targeted protein ions in the first quadrupole (Q1)
- Transferring isolated ions to the linear ion trap reaction cell (q2)
- Generating reagent ions via nano-ESI under denaturing conditions
- Isolating reagent ions in Q1 and transferring them to q2
- Mutually storing both ion populations in q2 for reaction times of 5-100 ms
- Mass-analyzing the products using time-of-flight detection [11] [13]
For ion attachment experiments, the reagent ion number density is varied by altering the voltage and injection time to optimize reactions [14]. Nitrogen gas is used in q2 at pressures ranging from 6-8 mtorr to facilitate collisions [13]. When analyzing large complexes like ribosome particles, the DC offsets between different regions are increased (e.g., 50-70 V between Q0 and q2) to collisionally activate the ions upon injection and drive off weakly-bound adduct species [13].
Charge Detection Mass Spectrometry (CDMS) Protocols
Charge detection mass spectrometry represents an alternative approach for analyzing macromolecules and heterogeneous complexes. CDMS enables direct measurement of both the m/z ratio and the charge of individual ions, circumventing the need for charge state resolution or deconvolution [17] [16]. Sample preparation for CDMS typically involves buffer exchange into volatile ammonium acetate solutions (e.g., 100 mM) using centrifugal filters or size exclusion chromatography columns [16]. For static nano-ESI direct infusion, proteins are diluted in 100 mM aqueous ammonium acetate to final concentrations of approximately 2 μM [16].
CDMS experiments are performed using specialized instrumentation, such as Q Exactive UHMR hybrid quadrupole Orbitrap mass spectrometers equipped with an ExD cell, which is autotuned for transmission of the specific proteins being analyzed [16]. Data acquisition times for CDMS typically range from several tens of minutes to hours to collect sufficient individual ion measurements for statistical significance [16]. Recent advances include the implementation of automatic ion control (AIC), which regulates ion flux based on signal density along the m/z axis rather than predefined injection times, facilitating the coupling of CDMS with liquid chromatography separation [16].
Analytical Applications in Macromolecular Analysis
Protein and Protein Complex Characterization
The generation of multiply charged ions has dramatically advanced the characterization of proteins and protein complexes. In native mass spectrometry, ESI under non-denaturing conditions preserves non-covalent interactions, enabling the study of protein-ligand, protein-protein, and protein-nucleic acid complexes [13]. The multiple charging phenomenon produces ions with relatively low m/z values despite their high molecular weights, making them amenable to analysis by conventional mass spectrometers. However, native conditions typically yield narrow charge state distributions, which can complicate mass measurement [13] [14].
Table 1: Multiply-Charged Ion Applications in Protein Analysis
| Application | Key Information | Representative Examples |
|---|---|---|
| Native MS | Preserves non-covalent complexes; narrow charge state distributions | Pyruvate kinase tetramer; Ferritin; GroEL [13] |
| Charge Inversion | Converts cations to anions via ion/ion reactions | Cytochrome c; Apo-myoglobin; Carbonic anhydrase [11] |
| Ion Attachment | Attaches ions of known mass/charge to determine unknown mass | E. coli ribosome particles; β-galactosidase [13] [14] |
| Charge Detection MS | Direct measurement of mass and charge for individual ions | Transferrin; β-galactosidase; AAVs; IgM oligomers [17] [16] |
The analysis of ribosomal particles exemplifies the challenges and solutions in macromolecular mass spectrometry. The E. coli 70S ribosome solution is typically prepared in buffers containing magnesium acetate to maintain structural integrity, followed by extensive buffer exchange (8 times) with 150 mM ammonium acetate and 10 mM magnesium acetate [14]. The inherent heterogeneity and extensive salt adduction result in significantly broadened peaks that are difficult to resolve using conventional approaches. The attachment of multiply-charged cations (e.g., the 10+ charge state of bovine ubiquitin or the 30+ charge state of bovine carbonic anhydrase) to these ribosomal anions has been shown to resolve multiple components, revealing particles with different combinations of missing components rather than intact 50S particles [14].
Analysis of Heterogeneous Biopharmaceuticals
Multiply charged ions have proven particularly valuable for characterizing heterogeneous biopharmaceuticals. Charge detection MS has been applied to complex molecules such as bispecific antibodies, antibody-drug conjugates (ADCs), gene therapies, and highly glycosylated proteins [17]. These analyses are challenging because traditional native MS requires either isotopic resolution or separation of charge states for mass determination, which becomes difficult with increasing sample heterogeneity [17] [16].
A prominent example is the analysis of the SARS-CoV-2 Spike protein, a trimeric glycosylated protein with potentially 8.2 × 10⁷⁵ possible glycoforms [17]. Traditional glycan analysis cannot provide information about how glycans at particular sites correlate with glycans at other sites on individual molecules. CDMS has demonstrated the capability to characterize this extreme heterogeneity and provide site-specific correlation information for glycans [17]. Similarly, CDMS has been used to characterize a complex mixture of IgM oligomers and co-occurring empty and genome-packed adeno-associated virus (AAV8) particles, where conventional native MS fails due to inability to resolve charge states in these heterogeneous mixtures [17].
Table 2: Mass Spectrometry Techniques for Macromolecular Analysis
| Technique | Key Features | Advantages | Limitations |
|---|---|---|---|
| Conventional ESI-MS | Multiple charging reduces m/z; Soft ionization | Broad mass range; Minimal fragmentation | Requires charge state resolution for mass determination |
| Ion/Ion Reactions | Charge manipulation via proton transfer or attachment | Charge inversion; Charge state simplification | Requires specialized instrumentation |
| Native MS | Preservation of non-covalent interactions | Study of intact complexes | Narrow charge state distributions; Salt adduction |
| Charge Detection MS | Simultaneous measurement of m/z and charge | No need for charge state resolution; Handles heterogeneity | Lower throughput; Complex data analysis |
Research Reagent Solutions and Essential Materials
Successful analysis of macromolecules using multiply charged ions requires specific reagents and materials optimized for different applications. The following table summarizes key research reagents and their functions in experimental workflows.
Table 3: Essential Research Reagents for Multiply-Charged Ion Experiments
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Hyaluronic Acids | Charge inversion reagents | HA with MW=1155 Da, 8-15k, 50k; HA-dp18 (MW=3412 Da) [11] |
| Protein Standards | Calibration and reagent ions | Cytochrome c; apo-myoglobin; carbonic anhydrase; ubiquitin [11] [14] |
| Ion/Ion Reagents | Multiply-charged ion attachment | Oxidized insulin chain A; holo-myoglobin with piperidine [13] [14] |
| MS Matrices | MALDI matrix compounds | CHCA; Cl-CCA; CHCA-C3; SA (sinapic acid) [12] |
| Buffers/Solvents | Sample preparation and LC mobile phases | Ammonium acetate; ammonium hydroxide; methanol; acetic acid [11] [16] |
| Nano-ESI Emitters | Ion source components | Borosilicate glass emitters [13] |
The choice of specific reagents depends on the experimental goals. For charge inversion experiments, hyaluronic acids of varying molecular weights serve as effective reagents for converting multiply charged protein cations to anions [11]. The relatively high charge densities of HA anions facilitate the extraction of multiple protons from proteins, leading to multiply charged protein anions [11]. For ion attachment applications, proteins such as oxidized insulin chain A (generating [IcA-5H]⁵⁻ and [IcA-6H]⁶⁻ ions) and holo-myoglobin prepared with piperidine serve as effective reagent anions [13]. The preparation of these reagents typically involves reconstituting lyophilized solids in appropriate solvents, with denaturing conditions (e.g., 50:50 H₂O:methanol with 1-5% glacial acetic acid) used to ensure higher charge state formation [14].
For MALDI applications, matrices with specific proton affinities are crucial for generating multiply charged ions. CHCA (PA = 841 kJ/mol) and Cl-CCA (PA = 838.5 kJ/mol) have been shown to produce higher charge states (up to +6 for myoglobin) compared to higher PA matrices like CHCA-C3 (PA = 879.5 kJ/mol) [12]. Homogeneous sample preparation using methods like forced dried droplet (FDD) is essential for reproducible multiply charged ion signals, with average RSD values of ~10-20% achieved for myoglobin samples with different matrices [12].
Visualization of Experimental Workflows
The analysis of macromolecules via multiply charged ions involves sophisticated instrumental setups and workflows. The following diagram illustrates a typical ion/ion reaction workflow for charge state manipulation and mass analysis.
The generation of multiply charged ions has fundamentally transformed the analysis of macromolecules, enabling mass spectrometric characterization of proteins, protein complexes, and other large biomolecules that were previously intractable. Electrospray ionization serves as the cornerstone technique for producing multiply charged ions, with ongoing advancements in ionization mechanisms, including novel charge inversion and ion attachment approaches. The experimental methodologies continue to evolve, providing researchers with powerful tools for investigating macromolecular structure and function. As mass spectrometry technology advances, the ability to generate and manipulate multiply charged ions will undoubtedly continue to expand the frontiers of macromolecular analysis, particularly for heterogeneous samples that challenge conventional analytical approaches. The integration of these techniques with separation methods and the development of specialized reagents and protocols will further enhance their application in both basic research and biopharmaceutical development.
Electrospray ionization (ESI), once primarily an analytical tool for mass spectrometry, has emerged as a platform for accelerated chemical synthesis. This transformation is driven by the recognition that microdroplets generated during ESI exhibit extraordinary reaction acceleration—up to a million-fold faster than conventional bulk-phase chemistry. This whitepaper explores the mechanistic basis for this phenomenon, focusing on the role of field ionization and chemical ionization in creating a unique reactive environment at microdroplet interfaces. The implications of these processes extend from fundamental understanding of ESI mechanisms to practical applications in pharmaceutical research, green chemistry, and prebiotic synthesis [18] [19].
From Analytical Tool to Synthetic Platform
Electrospray ionization mass spectrometry (ESI-MS) has traditionally served as a technique for transferring analytes from solution to the gas phase for mass analysis. Over the past decade, this role has expanded dramatically with the observation that microdroplets themselves function as microscopic reactors where chemical transformations occur at dramatically enhanced rates. This discovery has bridged the domains of chemical analysis and synthesis, enabling reactions that proceed rapidly without catalysts or pH adjustment under ambient conditions [18].
The significance of microdroplet chemistry lies in its dual utility: it provides a mechanism for ionization in ESI-MS while simultaneously facilitating rapid chemical synthesis. This dual function has prompted reevaluation of fundamental ESI mechanisms and opened new avenues for synthetic chemistry, particularly in contexts requiring rapid reaction screening or minimal reagent consumption [19].
Distinctive Features of Microdroplet Reactions
Several characteristic features distinguish microdroplet chemistry from conventional solution-phase reactions:
- Reaction acceleration up to 10⁶ times compared to bulk-phase analogs
- Interfacial phenomenon with demonstrated size-dependent effects
- Water dependency with minimal acceleration observed in completely dry solvents
- Strong electric fields at the air/water interface measuring ~10⁹ V/m
- Incomplete solvation of reagents reducing activation barriers
- Broad reaction scope including redox, condensation, hydrolysis, and substitution reactions
- Green chemistry attributes with reactions typically occurring without catalysts under ambient conditions [19]
These distinctive characteristics have been established through empirical investigation, but a coherent mechanistic explanation accounting for both the enhanced rates and diverse reaction types has remained elusive until recently.
Proposed Mechanism: Field Ionization and Chemical Ionization
Core Conceptual Framework
The proposed mechanism unifying diverse observations in microdroplet chemistry centers on two sequential processes: field ionization (FI) followed by chemical ionization (CI). This mechanism explains both molecular ionization for mass spectrometric detection and the dramatically accelerated reaction kinetics observed in microdroplets [19].
The model incorporates three key concepts:
Partial Solvation: Interfacial species experience incomplete solvation, destabilizing reagents more than transition states and thereby increasing reaction rate constants [19].
Strong Electric Fields: Microdroplet interfaces containing water maintain intrinsic electric fields of ~10⁹ V/m, as established through computational and experimental measurements [19].
Reactive Species Generation: Field ionization of water creates primary reactive species (H₂O⁺˙) that initiate cascades of chemical transformations [19].
Field Ionization Step
The strong electric field at microdroplet interfaces enables field ionization of water molecules through electron tunneling, analogous to Beckey's field ionization technique. This process generates water radical cations (H₂O⁺˙) as the primary oxidizing agents and solvated electrons (e⁻(aq)) as the primary reducing agents [19].
Field ionization occurs with effectively zero activation energy due to the quantum mechanical nature of electron tunneling. The probability of tunneling depends on both molecular orientation and electric field strength across the interfacial water layer. Notably, electron tunneling exhibits lower effective energy barriers in aqueous environments compared to vacuum, enhancing its probability at microdroplet interfaces [19].
Chemical Ionization Step
Following field ionization, the initially generated reactive species undergo secondary reactions in a chemical ionization process. The water radical cation reacts with neutral water molecules to form a dimeric complex that dissociates into hydronium ion and hydroxyl radical:
Equation 1: Self-Chemical Ionization H₂O⁺˙ + H₂O → H₂O⁺˙(H₂O) → H₃O⁺ + HO˙ [19]
This "self-chemical ionization" represents a crucial step in generating the reactive intermediates responsible for diverse microdroplet transformations. The hydroxyl radical and hydronium ion subsequently participate in acid-base and redox chemistry, while the solvated electron serves as a potent reducing agent [19].
Table 1: Reactive Species in Microdroplet Chemistry
| Species | Formation Process | Chemical Role | Representative Reactions |
|---|---|---|---|
| H₂O⁺˙ | Primary field ionization | Strong oxidizing agent | Electron transfer reactions |
| e⁻(aq) | Electron from FI | Strong reducing agent | Reduction of organic compounds |
| HO˙ | Secondary CI from H₂O⁺˙ | Oxidizing agent | Hydrogen abstraction, addition |
| H₃O⁺ | Secondary CI from H₂O⁺˙ | Brønsted acid | Acid-catalyzed reactions |
Mechanistic Integration in ESI Process
The integration of FI and CI mechanisms provides a comprehensive explanation for microdroplet phenomena. This unified model accounts for:
- Ionization for MS detection through protonation and redox processes
- Reaction acceleration via highly reactive radical and ionic intermediates
- Simultaneous acid-base and redox chemistry within single droplet populations
- Generation of observed products including hydrogen peroxide through secondary reactions [19]
The mechanism operates specifically at the air-water interface of microdroplets where both strong electric fields and partial solvation conditions prevail, explaining the interfacial nature of reaction acceleration.
Experimental Evidence and Validation
Key Experimental Findings
Multiple lines of evidence support the proposed FI/CI mechanism in microdroplets:
Electric field measurements: Experimental determinations confirm field strengths sufficient for field ionization (~10⁹ V/m) at nebulized microdroplet interfaces [19].
Water radical cation detection: Mass spectrometric identification of H₂O⁺˙ and its oligomers (m/z 36) provides direct evidence for the primary FI step [19].
Size-dependent effects: Correlation between droplet size (and thus surface-to-volume ratio) and reaction acceleration confirms the interfacial nature of the process [19].
Product analysis: Reaction products consistent with radical and acid-base mechanisms support the proposed reactive intermediates [19].
Experimental Approaches for Microdroplet Analysis
Researchers have employed diverse methodological approaches to investigate microdroplet chemistry:
Table 2: Experimental Methods in Microdroplet Research
| Method | Key Features | Applications | References |
|---|---|---|---|
| ESI-MS | Online reaction monitoring | Real-time analysis of reaction kinetics and products | [18] |
| Levitated Droplets | Single droplet manipulation | Controlled study of interfacial phenomena | [19] |
| Spray Collection | Product isolation | Milligram-scale synthesis for NMR characterization | [19] |
| Computational Modeling | Theoretical simulation | Electric field calculation and reaction pathway analysis | [19] |
| Imprint DESI-MSI | Spatial visualization | Herbicide penetration in plant tissues | [20] |
Quantitative Data on Reaction Acceleration
Microdroplet environments enhance reaction rates by several orders of magnitude compared to bulk phase conditions. The following table summarizes documented acceleration factors for various reaction types:
Table 3: Quantitative Acceleration Factors in Microdroplet Reactions
| Reaction Type | Acceleration Factor | Key Characteristics | Required Conditions | |
|---|---|---|---|---|
| Claisen-Schmidt | Up to 10⁶ | Catalyst-free condensation | Aqueous microdroplets, ambient temperature | [19] |
| Pomeranz-Fritsch | Up to 10⁶ | Cyclization reaction | Interfacial environment, strong electric field | [19] |
| Redox Reactions | 10³-10⁵ | Diverse substrates | Water-containing droplets, partial solvation | [19] |
| Acid-Base Catalyzed | 10⁴-10⁶ | No external acid/base | H₃O⁺/HO˙ generation at interface | [19] |
| Hydrolysis | 10³-10⁵ | Spontaneous cleavage | Interfacial water activation | [19] |
Methodologies for Microdroplet Experiments
Standard ESI-MS Protocol for Reaction Monitoring
Principle: Electrospray ionization generates microdroplets while simultaneously serving as an inlet for mass spectrometric analysis [19].
Procedure:
- Prepare reagent solution in water-containing solvent (typically 1-100 µM concentration)
- Load solution into ESI source with flow rates of 0.5-5 µL/min
- Apply high voltage (3-6 kV) to create charged microdroplets
- Monitor reaction products in real-time using mass analyzer
- Optimize desolvation temperature and gas flows to control droplet lifetime
Key Parameters:
- Voltage: 3-6 kV
- Flow rate: 0.5-5 µL/min
- Solvent: Water-containing (often 1:1 to 4:1 organic:water)
- Capillary temperature: 150-300°C
Applications: Rapid reaction screening, mechanistic studies, catalyst-free synthesis [19].
Spray Collection for Product Isolation
Principle: Microdroplets are generated through spraying and collected for offline analysis, enabling product isolation in milligram quantities [19].
Procedure:
- Prepare reagent solution (mM concentration)
- Nebulize or electrospray solution toward collector surface
- Control droplet flight distance to manipulate reaction time
- Collect products in solvent or on solid surface
- Characterize using NMR, LC-MS, or other analytical techniques
Key Parameters:
- Nozzle-to-collector distance: 1-10 cm
- Solution concentration: 1-100 mM
- Collection solvent: Compatible with product solubility
- Gas pressure: 20-120 psi (for nebulization)
Applications: Small-scale synthesis, product characterization, reference standard preparation [19].
Imprint DESI-MSI for Spatial Analysis
Principle: Imprint desorption electrospray ionization mass spectrometry imaging enables visualization of spatial distributions in complex samples [20].
Procedure:
- Treat biological sample (plant/animal tissue) with analyte of interest
- Section sample and press against oil-absorbing film for 10 seconds
- Analyze imprint using DESI-MSI with raster scanning
- Acquire mass spectra at each pixel (150 µm resolution)
- Reconstruct spatial distribution using custom software
Key Parameters:
- Spray voltage: -6 kV (negative mode)
- Solvent: 8:2 acetonitrile:water
- Flow rate: 1.5 µL/min
- Pixel size: 150 µm
- Gas pressure: 120 psi
Applications: Herbicide penetration studies, metabolite localization, drug distribution analysis [20].
Visualizing Microdroplet Mechanisms and Workflows
FI/CI Mechanism in Microdroplets
Microdroplet FI/CI Mechanism Diagram: This diagram illustrates the sequential process of field ionization followed by chemical ionization that generates reactive species in charged microdroplets.
Microdroplet Experiment Workflow
Microdroplet Experiment Workflow: This workflow outlines the typical steps in microdroplet experiments, from sample preparation to final applications.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Materials for Microdroplet Experiments
| Item | Specifications | Function | Example Sources/References |
|---|---|---|---|
| High-Voltage Power Supply | 3-6 kV capability | Electrospray droplet generation | Standard ESI instrumentation [19] |
| Microsyringe Pump | 0.5-5 µL/min flow range | Precise solution delivery | Commercial HPLC systems [19] |
| Water-Soluble Reagents | High purity (>95%) | Reaction substrates | Sigma-Aldrich, Fisher Scientific [19] |
| Mass Spectrometer | High resolution (>30,000) | Reaction monitoring and product identification | Orbitrap platforms [20] |
| Oil-Absorbing Film | Clean & Clear or equivalent | Imprint substrate for DESI-MSI | Johnson & Johnson [20] |
| Nebulization Gas | High-purity N₂ (99.999%) | Droplet generation without ESI | Standard laboratory gas supply [19] |
| Alkali Metal Salts | CsI for polymer stability | Adduct formation for MS analysis | Chemical suppliers [21] |
Implications and Applications
Implications for ESI Mechanism Understanding
The FI/CI model revolutionizes understanding of fundamental ESI processes by:
- Providing a unified mechanism for both ionization and reaction acceleration
- Explaining the essential role of water in ESI processes
- Accounting for the generation of protonated molecules without external acid sources
- Revealing the connection between electric fields and reactive intermediate generation [19]
This enhanced understanding enables more intentional application of ESI not just as an analytical tool but as a synthetic platform.
Practical Applications Across Industries
Drug Discovery: Microdroplet acceleration enables rapid screening of reaction pathways and metabolite identification, significantly reducing development timelines. The ability to perform catalyst-free reactions under ambient conditions provides sustainable synthetic routes for pharmaceutical intermediates [18] [19].
Green Synthesis: The elimination of requirement for toxic catalysts and harsh conditions aligns with green chemistry principles. Microdroplet approaches reduce solvent consumption and energy requirements while maintaining high reaction efficiency [18].
Prebiotic Chemistry: The demonstration that complex organic transformations can occur spontaneously in aqueous microdroplets under ambient conditions has implications for understanding chemical evolution and the origin of life [18] [19].
Analytical Sciences: Improved understanding of ESI mechanisms enhances interpretation of mass spectrometric data, particularly for labile compounds and reaction intermediates. The application of imprint DESI-MSI extends to food safety monitoring and environmental toxicology [20].
The integration of field ionization and chemical ionization mechanisms provides a comprehensive framework for understanding the unique chemistry occurring in charged microdroplets. This model explains both the remarkable acceleration of diverse reactions and the ionization processes fundamental to ESI-MS. As research in this field advances, the deliberate exploitation of microdroplet environments promises to transform approaches to chemical synthesis, analysis, and understanding of fundamental chemical processes. The implications span from practical applications in pharmaceutical research to fundamental questions about chemical reactivity at interfaces.
ESI-MS in Action: Transformative Applications in Clinical and Pharmaceutical Laboratories
Electrospray Ionization Tandem Mass Spectrometry (ESI-Tandem-MS or ESI-MS/MS) represents a cornerstone technological advancement in clinical chemistry, revolutionizing the screening and diagnosis of Inborn Errors of Metabolism (IEM). IEM constitute a group of phenotypically and genotypically heterogeneous metabolic disorders caused by gene mutations encoding metabolic pathway enzymes or receptors. Deficiency or changes in the activity of essential enzymes in intermediate metabolic pathways lead to the accumulation or deficiency of corresponding metabolites, manifesting in a wide range of diseases with clinical heterogeneity that complicates their diagnosis [22].
The application of ESI-MS/MS to newborn screening offers the potential of substantially altering the natural history of these diseases by reducing morbidity and mortality. This technology has transitioned screening from the outdated classical methods of "one test, one metabolite, and one disease" to a "single test, many metabolites, and many diseases" approach [22]. The ability to detect numerous metabolic disorders simultaneously with high sensitivity and specificity has established ESI-MS/MS as an ethical, safe, simple, and reliable screening test that is now a mandatory public health strategy in most developed countries [22].
Fundamental Principles of Electrospray Ionization
ESI Mechanism and Process
Electrospray Ionization (ESI) is a soft ionization technique that operates at atmospheric pressure and is particularly suitable for thermally labile and at least moderately polar organic analytes. The ESI process involves three fundamental stages:
Droplet Formation: A liquid sample is pumped through a narrow capillary needle maintained at high voltage (typically 3-5 kV), creating a fine aerosol of charged droplets.
Droplet Desolvation: The charged droplets shrink through solvent evaporation, increasing charge density until Coulombic repulsion overcomes surface tension.
Ion Emission: Gaseous ions are released via two possible mechanisms - the charged residue model (CRM) for larger molecules or the ion evaporation model (IEM) for smaller ions [23].
ESI typically yields ions with no unpaired electrons (even-electron ions), with the resulting [M + H]+ species referred to as protonated molecules. These ions possess low internal energy, resulting in minimal fragmentation in single-stage MS experiments, making ESI ideal for preserving intact molecular information for sensitive and fragile compounds [23].
ESI in Positive and Negative Ion Modes
The versatility of ESI allows operation in both positive (ES+) and negative (ES-) ion modes, with mode selection dependent on the proton affinity of the target analytes:
ES+ Mode: Optimal for compounds with basic characteristics, easily ionized via adduct formation with proton(s). Common adducts include [M + H]+, [M + Na]+, and [M + NH4]+.
ES- Mode: Suitable for molecules possessing acidic functional groups and lacking basic ones, typically forming [M - H]- ions [23].
The selection of ionization mode significantly impacts detection sensitivity and should be optimized based on the chemical properties of target metabolites. For IEM screening, positive ion mode is typically employed for amino acids and acylcarnitines, though some applications may benefit from negative mode detection.
ESI-MS/MS Methodology for IEM Screening
Analytical Workflow
The ESI-MS/MS screening process for IEM follows a standardized workflow that ensures comprehensive metabolite profiling with high reproducibility:
Sample Preparation: Heel blood from newborns (3 days after birth) is collected and dripped onto filter paper (S&S 903), dried naturally at room temperature, and extracted for analysis [24].
Chromatographic Separation: While Flow Injection Analysis (FIA) can be used for high-throughput screening, Liquid Chromatography (LC) provides superior separation of isomeric and isobaric compounds. Typical LC conditions utilize C18 columns with mobile phases consisting of methanol/water mixtures, often with 0.01% formic acid to enhance protonation [25].
Mass Spectrometric Analysis: The instrument setup for IEM screening typically includes:
- Ion Source: ESI with interface voltage of 4.5 kV
- Nebulizer Gas Flow: 3 L/min
- Drying Gas Flow: 15 L/min
- Desolvation Line Temperature: 250°C
- Heat Block Temperature: 400°C [25]
Data Analysis: Quantitative analysis of amino acids and acylcarnitines using multiple reaction monitoring (MRM) transitions specific to each metabolite.
Key Metabolites and Disease Associations
ESI-MS/MS enables simultaneous monitoring of numerous metabolites that serve as biomarkers for various IEM categories. The table below summarizes the primary metabolite classes and their associated disorder groups:
Table 1: Key Metabolite Classes and Associated IEM Categories
| Metabolite Class | Representative Analytes | Associated Disorder Categories | Clinical Significance |
|---|---|---|---|
| Amino Acids | Phenylalanine, Tyrosine, Leucine, Methionine | Aminoacidemias (e.g., PKU, MSUD, HCY) | Accumulation of toxic amino acids causes neurological damage |
| Acylcarnitines | C0, C2, C3, C4, C5, C8, C16 | Organic Acidemias, Fatty Acid Oxidation Disorders | Indicators of blocked metabolic pathways and energy deficiency |
| Succinylacetone | - | Tyrosinemia Type I | Liver and kidney dysfunction |
The sensitivity and specificity of this method can reach 99% and 99.995%, respectively, for most amino acid disorders, organic acidemias, and fatty acid oxidation defects [22].
Experimental Protocols for IEM Screening
Standardized Newborn Screening Protocol
Materials and Reagents:
- Filter paper cards (S&S 903)
- Internal standards for amino acids and acylcarnitines
- Methanol (HPLC grade)
- Formic acid (analytical grade)
- Butanol/HCl derivatization reagent (for some methods)
Procedure:
- Collect heel-prick blood spots on filter paper 24-48 hours after birth
- Dry blood spots at ambient temperature for a minimum of 3 hours
- Punch 3.2 mm discs from dried blood spots into microtiter plates
- Add internal standard solution containing deuterated amino acids and acylcarnitines
- Extract analytes with 100 μL methanol with shaking for 30 minutes
- Derivatize butyl esters (for some methods) or analyze underivatized
- Analyze using LC-ESI-MS/MS with a run time of approximately 2 minutes per sample
- Quantify metabolites against calibration curves using stable isotope internal standards
Quality Control:
- Include quality control samples with each batch (low, medium, high concentrations)
- Monitor internal standard responses for signal suppression/enhancement
- Participate in external proficiency testing programs
Two-Tier Screening Approach
To minimize false positives and improve specificity, a two-tier screening system is often implemented:
- First-Tier Screening: Initial FIA-MS/MS analysis for high-throughput screening
- Second-Tier Testing: Confirmatory LC-MS/MS analysis for presumptive positive samples, increasing test specificity and dramatically reducing false positives [22]
This approach is particularly valuable for disorders with low specificity biomarkers, where second-tier tests can utilize different chromatographic separations or additional analyte markers to improve positive predictive value.
Table 2: Incidence Rates of Common IEM Detected by ESI-MS/MS in Southern China (n=111,986 newborns)
| Disorder | Incidence Rate | Primary Metabolic Markers | Confirmatory Testing |
|---|---|---|---|
| Primary Carnitine Deficiency | 1:9,332 | Decreased free carnitine (C0) | SLC22A5 gene sequencing |
| Phenylketonuria (PKU) | 1:18,664 | Elevated phenylalanine | PAH gene analysis |
| Isovaleric Acidemia | 1:37,329 | Elevated C5 acylcarnitine | IVD gene sequencing |
| Citrullinemia Type II | 1:111,986 | Elevated citrulline | SLC25A13 mutation analysis |
| Methylmalonic Acidemia | 1:37,329 | Elevated C3 acylcarnitine | MMUT, MMAA, MMAB genes |
| Overall IEM Incidence | 1:3,733 | Multiple markers | Gene sequencing |
Data adapted from a screening study of 111,986 newborns in Southern China [24].
Comparative Analysis with Alternative Ionization Techniques
ESI versus APCI for IEM Applications
While ESI is the predominant ionization source for IEM screening, Atmospheric Pressure Chemical Ionization (APCI) represents an alternative approach with distinct characteristics:
Table 3: Comparison of ESI and APCI Characteristics for Clinical Applications
| Parameter | Electrospray Ionization (ESI) | Atmospheric Pressure Chemical Ionization (APCI) |
|---|---|---|
| Ionization Mechanism | Charge transfer in liquid phase | Gas-phase chemical ionization |
| Analyte Polarity | Moderate to high polarity | Low to moderate polarity |
| Molecular Mass Range | Up to 100,000+ Da | < 1,500 Da |
| Matrix Effects | More susceptible to suppression | Less susceptible to matrix effects |
| Flow Rate Compatibility | Optimal at low flow rates (<0.2 mL/min) | Compatible with higher flow rates (1.0 mL/min) |
| Adduct Formation | Pronounced ([M+H]+, [M+Na]+, etc.) | Primarily [M+H]+ or [M-H]- |
| Fragmentation Pattern | Even-electron ions, heterolytic cleavage | Can generate odd-electron ions |
APCI appears to be slightly less liable to matrix effects than ESI, as demonstrated in studies comparing the ionization sources for pharmaceutical compounds [25]. However, ESI generally provides superior sensitivity for polar metabolites central to IEM screening.
Advancing Ionization Technologies
Beyond conventional ESI, several advanced ionization techniques have emerged with potential applications in specialized IEM testing:
Desorption Electrospray Ionization (DESI):
- Ambient ionization technique requiring minimal sample preparation
- Utilizes charged solvent spray to desorb and ionize analytes directly from surfaces
- Enables spatial mapping of metabolites in tissue samples [26]
Nanospray Desorption Electrospray Ionization (nanoDESI):
- Improved version achieving spatial resolution of ~10 μm
- Enhanced sensitivity through better ion transfer efficiency
- Suitable for gentle surface lipidomic mapping [26]
These ambient ionization methods expand the application of MS-based metabolic profiling beyond dried blood spots to tissue sections and other complex samples.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of ESI-MS/MS for IEM screening requires carefully selected reagents and materials optimized for metabolic profiling:
Table 4: Essential Research Reagents for ESI-MS/MS IEM Screening
| Reagent/Material | Function | Technical Specifications | Application Notes |
|---|---|---|---|
| Filter Paper Cards | Blood sample collection and storage | S&S 903 specification | Ensure uniform blood saturation and drying |
| Deuterated Internal Standards | Quantitative calibration | Isotope-labeled amino acids and acylcarnitines | Correct for matrix effects and ionization efficiency |
| Methanol (HPLC Grade) | Protein precipitation and extraction | LC-MS grade, low UV absorbance | Minimize background interference in MS analysis |
| Formic Acid | Mobile phase additive | ≥99% purity, LC-MS compatible | Enhances protonation in positive ion mode (0.01-0.1%) |
| Acylcarnitine Standards | Calibration and quality control | Certified reference materials | Multi-point calibration for quantitative accuracy |
| Amino Acid Standards | Calibration and quality control | Certified reference materials | Cover essential amino acids relevant to IEM |
Data Interpretation and Clinical Correlation
Diagnostic Algorithms
Interpretation of ESI-MS/MS data for IEM screening follows established algorithms that integrate multiple metabolite ratios and absolute concentrations:
Primary Marker Elevation: Identify significantly elevated primary biomarkers (e.g., phenylalanine for PKU)
Ratio Analysis: Calculate diagnostically relevant ratios (e.g., phenylalanine/tyrosine for PKU confirmation)
Pattern Recognition: Identify characteristic acylcarnitine profiles (e.g., elevated C0 for carnitine uptake defect)
Dynamic Monitoring: Track changes in metabolite levels in response to treatment
The integration of computational tools and bioinformatics pipelines has significantly enhanced the accuracy and efficiency of data interpretation in large-scale screening programs.
Integration with Genetic Confirmation
ESI-MS/MS serves as a biochemical screening tool that is increasingly integrated with genetic confirmation:
Next-generation sequencing (NGS) is now included in confirmatory testing in many countries. Genomic DNA isolated from dried blood spots can be used for NGS, providing reliable sequencing results as a secondary diagnostic test for NBS [22].
Future Perspectives and Research Directions
The field of ESI-MS/MS applications in IEM screening continues to evolve with several emerging trends:
Expanded Disorder Panels: Continuous addition of new disorders to screening panels as biomarkers are validated
Second-Tier Molecular Testing: Integration of genetic testing directly into the screening algorithm
Computational Advancements: Improved bioinformatics tools for data interpretation and variant prioritization
International Standardization: Harmonization of cutoff values and analytical protocols across screening programs
Novel Biomarker Discovery: Application of untargeted metabolomics to identify new IEM biomarkers
According to bibliometric analysis of the field, the most relevant current research directions include "selective screening for IEM," "new treatments for IEM," "new disorders considered for MS/MS testing," "ethical issues associated with newborn screening," "new technologies that may be used for newborn screening," and "use of a combination of MS/MS and gene sequencing" [22].
The reproducibility and automation of metabolome annotation are being enhanced through workflows like MAW (Metabolome Annotation Workflow), which combines MS2 data pre-processing, spectral and compound database matching with computational classification, and in silico annotation [27]. Such computational advances are crucial for handling the increasing complexity of metabolomics data in IEM screening.
ESI-Tandem-MS has fundamentally transformed the landscape of IEM screening, enabling early detection of metabolic disorders before symptom onset and significantly improving clinical outcomes through timely intervention. The technology's unparalleled sensitivity, specificity, and multiplexing capacity have established it as the gold standard in newborn screening programs worldwide.
Ongoing advancements in ionization techniques, computational analytics, and integration with genomic technologies promise to further enhance the scope and accuracy of IEM detection. As the field progresses, ESI-MS/MS will continue to play a pivotal role in advancing preventive medicine and reducing the global burden of inherited metabolic disorders through early intervention and personalized treatment approaches.
The structural elucidation of hemoglobin variants represents a critical endeavor in clinical proteomics, essential for diagnosing hemoglobinopathies—among the most common inherited human disorders globally [28]. Over 1,000 hemoglobin variants have been characterized, with approximately 150 causing clinically significant conditions such as sickle cell anemia, hemolytic anemia, and thalassemias [28]. The precision of variant identification directly impacts medical care, prognosis, and genetic counseling outcomes. Electrospray Ionization (ESI) Mass Spectrometry has revolutionized this field by enabling rapid, accurate analysis of hemoglobin proteins with minimal sample requirements. This technical guide explores ESI-based methodologies within the broader context of ionization mechanism research, providing researchers and drug development professionals with advanced protocols for characterizing variant hemoglobins. The exceptional capability of ESI to generate multiply-charged ions from large biomolecules like hemoglobin (approximately 64.5 kDa) has transformed mass spectrometry from merely an analytical tool into a comprehensive platform for both structural analysis and accelerated chemical synthesis [18].
Electrospray Ionization Fundamentals and Mechanisms
Electrospray Ionization operates through a sophisticated mechanism that converts solution-phase proteins into gas-phase ions amenable to mass analysis. When applied to hemoglobin analysis, the ESI process begins with introducing a hemoglobin solution through a capillary maintained at high voltage (typically 3-4 kV). This creates a Taylor cone that emits a fine mist of charged droplets toward the mass spectrometer inlet [29]. As these droplets travel through the ESI source, solvent evaporation occurs while droplet charge density increases. Through processes termed field ionization and chemical ionization, the droplets eventually release protonated hemoglobin molecules into the gas phase [18].
Recent research on charged microdroplets has revealed that ESI facilitates remarkably accelerated chemical reactions—up to 10^6 times faster than analogous bulk reactions—which has profound implications for both analytical applications and synthetic chemistry [18]. This phenomenon stems from unique ionization mechanisms within microdroplets, including extremely high electric fields at the droplet surface that promote efficient ionization. For hemoglobin analysis, this translates to enhanced sensitivity and the ability to detect low-abundance variants that might escape conventional methodologies.
A key advantage of ESI for hemoglobin analysis is the generation of multiply-charged ions [M+nH]ⁿ⁺, which effectively reduces the mass-to-charge ratio (m/z) of large proteins like hemoglobin, making them compatible with most mass analyzers. The number of charges acquired depends on solvent composition, source parameters, and the number of accessible basic sites on the protein surface. This multi-charging phenomenon enables precise molecular weight determination of intact globin chains and facilitates top-down sequencing approaches through tandem mass spectrometry.
Analytical Approaches for Hemoglobin Variant Characterization
Mass Spectrometry Platforms and Methodologies
Multiple mass spectrometry platforms have been successfully employed for hemoglobin variant analysis, each offering distinct advantages:
Electrospray Ionization Mass Spectrometry (ESI-MS) provides excellent capabilities for molecular weight determination of intact globin chains. The methodology requires minimal sample preparation (as little as 2-10 μL of whole blood) with no need for pre-separation of red cells or globin chains [30]. Aged and hemolyzed blood samples can also be analyzed effectively. ESI-MS typically achieves positive identification of 95% of variants, with a sample preparation and analysis time of approximately one hour [30]. This approach has successfully identified 99 different abnormalities comprising 36 alpha-chain variants, 59 beta-chain variants, and 4 hybrid hemoglobins, including novel variants [30].
Liquid Chromatography-High Resolution Mass Spectrometry (LC-HR-MS) combines separation power with mass accuracy to resolve challenging variants. Using a C4 reversed-phase column, this method can separate pairs of normal and variant Hb subunits with mass differences smaller than 1 Da [31]. The high resolution enables explicit observation of analytes in deconvoluted MS1 mass spectra and facilitates top-down analysis for matching amino acid sequences to correct Hb variant subunits [31].
MALDI-ISD Mass Spectrometry utilizes matrix-assisted laser desorption/ionization with in-source decay for top-down sequencing. With appropriate matrix selection (super DHB or 1,5-diaminonaphthalene), this technique provides extensive fragmentation generating c-, (z+2)-, and y-ion series [28]. On the first 70 amino acids from the N- and C-termini of alpha and beta chains can be covered in a single experiment, enabling characterization of variants like Hb Westmead (α122 His→Gln) [28].
Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry (ESI-ETD-MS) excels at fragmenting multiply-charged ions (3+ or higher) to generate sequence information. This approach is particularly valuable when variant peptide ions experience interference from other ions at the same m/z value [29]. ETD spectra are often less complex and easier to interpret than collision-induced dissociation spectra of the same charge state [29].
Comparative Performance of Analytical Methods
Table 1: Comparison of Mass Spectrometry Methods for Hemoglobin Variant Analysis
| Method | Key Features | Analysis Time | Mass Accuracy | Sequence Coverage | Key Applications |
|---|---|---|---|---|---|
| ESI-MS | Minimal sample prep; intact mass measurement | ~1-2 hours [30] | Moderate | N/A | Rapid screening; molecular weight profiling [30] |
| LC-HR-MS | High resolution; separation of similar mass variants | Moderate-long | High (<1 Da difference) [31] | Limited without fragmentation | Distinguishing variants with mass differences <1 Da [31] |
| MALDI-ISD MS | Top-down sequencing; extensive fragmentation | Rapid | High | ~70 N- and C-terminal residues [28] | Variant localization; diagnostic marker identification [28] |
| ESI-ETD MS | Fragmentation of high charge-state ions; simple spectra | Moderate | High | Dependent on charge state | Interference resolution; variant confirmation [29] |
Experimental Protocols
ESI-MS Analysis of Intact Hemoglobin Variants
Sample Preparation:
- Dilute whole blood samples 50-fold with deionized water [29]
- Denature using organic solvents (e.g., 50% aqueous acetonitrile) with acidification (0.2% formic acid) to promote ionization [29]
- Centrifuge if necessary to remove particulate matter
Instrument Parameters (SYNAPT G2 System):
- Ionization mode: ESI positive [29]
- Capillary voltage: 3.5 kV [29]
- Cone voltage: 30 V [29]
- Desolvation temperature: 200°C [29]
- Source temperature: 120°C [29]
- Infusion flow rate: 5 μL/min [29]
Data Interpretation:
- Compare measured molecular weights of α-globin (15,126 Da) and β-globin (15,867 Da) chains to theoretical values
- Mass shifts indicate specific amino acid substitutions (e.g., +30 Da suggests Ala→Thr, Arg→Trp, Gly→Ser, Thr→Met, or Val→Glu) [29]
- Reference database of known variants to narrow possibilities
ESI-ETD MS for Variant Sequencing
Sample Digestion:
- Dilute blood sample 50-fold with water and denature [29]
- Digest with trypsin for 30 minutes [29]
- Further dilute digest 10-fold with 50% aqueous acetonitrile containing 0.2% formic acid [29]
ETD Parameters:
- Reagent: para-nitrotrotoluene (m/z 137) [29]
- Glow discharge current: 55 μA (negative mode) [29]
- Helium bath gas pressure: 5 × 10⁻² mbar [29]
- Transfer T-Wave cell pressure: 5 × 10⁻³ mbar with argon [29]
- T-Wave speed and amplitude: 300 m/sec and 0.2 V respectively [29]
Data Analysis:
- Identify variant peptides through mass anomalies in MS1 spectrum
- Select triply-charged precursor ions for ETD fragmentation
- Sequence fragment ions to locate exact mutation site
- Confirm variant identity through characteristic fragment mass shifts
Table 2: Common Hemoglobin Variants and Their Mass Characteristics
| Variant | Amino Acid Substitution | Theoretical Mass Shift (Da) | Clinical Significance | Prevalence |
|---|---|---|---|---|
| Hb S | β6 Glu→Val [28] | -30 (β-chain) | Sickle cell anemia | Up to 8% trait in African Americans [28] |
| Hb C | β6 Glu→Lys [28] | +1 (β-chain) | Mild hemolytic anemia | Up to 2% trait in African Americans [28] |
| Hb E | β26 Glu→Lys [28] | +1 (β-chain) | Mild microcytosis | 30-60% in Southeast Asia [28] |
| Hb D-Punjab | β121 Glu→Gln [28] | +1 (β-chain) | Generally benign | 3% in India and Pakistan [28] |
| Hb Westmead | α122 His→Gln [28] | +1 (α-chain) | Generally benign | Rare |
Top-Down LC-HR-MS for Intact Subunit Analysis
Chromatographic Conditions:
- Column: C4 reversed-phase [31]
- Mobile phase: Gradient of water/acetonitrile with 0.1% formic acid
- Flow rate: Optimized for subunit separation
Mass Spectrometry Parameters:
- Resolution: >30,000 for clear subunit separation
- Scan range: m/z 800-2000 for multiply-charged ions
- Deconvolution algorithm: Maximum entropy or similar for intact mass determination
Variant Identification:
- Compare retention times of α- and β-globin chains to controls
- Deconvolute mass spectra to determine intact protein mass
- Identify mass shifts indicative of specific variants
- For ambiguous identifications, employ top-down fragmentation
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Hemoglobin Variant Analysis
| Reagent/Material | Function | Application Example |
|---|---|---|
| C4 Reversed-Phase Columns | Separation of intact globin subunits | LC-HR-MS analysis of variants with minimal mass differences [31] |
| Trypsin (Proteomic Grade) | Enzymatic digestion for bottom-up analysis | Generation of peptides for variant identification by ESI-ETD MS [29] |
| para-Nitrotoluene | ETD reagent anion source | Electron transfer dissociation for sequencing multiply-charged peptides [29] |
| Super DHB Matrix | MALDI matrix with high ISD efficiency | In-source decay fragmentation for top-down sequencing of hemoglobin variants [28] |
| Formic Acid | Mobile phase modifier; protein denaturant | Improving ionization efficiency in ESI-based methods [29] |
| Acetonitrile (HPLC Grade) | Organic mobile phase component | Reversed-phase separation of hemoglobin subunits and peptides [29] |
Workflow Visualization
Diagram 1: Comprehensive Workflow for Hemoglobin Variant Analysis using ESI-Based Methods
The field of hemoglobin variant analysis continues to evolve with advancements in ESI technology and methodology. Recent research into charged microdroplet chemistry suggests potential for further acceleration of analysis times and enhanced detection sensitivity [18]. The observation of reaction acceleration up to 10^6 times in microdroplets compared to bulk solutions opens new avenues for rapid sample preparation and potentially novel ionization approaches [18].
The integration of ion mobility spectrometry with ESI-MS provides an additional dimension of separation based on protein shape and size, potentially resolving variants with identical masses but different structures [29]. This approach is particularly valuable for detecting post-translational modifications and structural hemoglobin variants that may not result in mass changes.
As mass spectrometry instrumentation becomes more accessible and user-friendly, ESI-based methods are transitioning from specialized reference laboratories to routine clinical practice. This democratization of advanced analytical capabilities promises to improve diagnosis and monitoring of hemoglobinopathies worldwide, particularly in regions with high prevalence of these genetic disorders.
In conclusion, ESI mass spectrometry has established itself as an indispensable tool for the structural elucidation of hemoglobin variants, offering unparalleled speed, accuracy, and versatility. The methodologies detailed in this technical guide provide researchers and clinical scientists with robust frameworks for implementing these powerful approaches in both research and diagnostic contexts. As our understanding of electrospray ionization mechanisms deepens, further refinements and innovations will undoubtedly enhance our ability to characterize hemoglobin variants and improve patient care for those affected by hemoglobinopathies.
Dissociation Constant (KD) Simply Explained
The dissociation constant (KD) serves as a fundamental quantitative parameter in drug discovery, measuring the strength of non-covalent interactions between protein targets and therapeutic ligands. Defined as the ligand concentration at which half of the protein's binding sites are occupied at equilibrium, KD provides critical insights into binding affinity, with lower values (typically picomolar to nanomolar) indicating tighter binding and higher values (micromolar and above) reflecting weaker interactions [32]. Accurate KD determination enables medicinal chemists to understand structure-activity relationships, optimize lead compounds, and rationally design drugs with enhanced potency and selectivity.
Within the broader context of electrospray ionization (ESI) research, the application of ESI-mass spectrometry (ESI-MS) has revolutionized the study of these non-covalent complexes. ESI-MS allows for the direct detection and characterization of protein-ligand interactions without requiring ligand labeling or immobilization, making it particularly valuable for fragment-based drug discovery and high-throughput screening [33] [34]. This technical guide explores established and emerging methodologies for KD determination, with particular emphasis on ESI-MS techniques and their integration within modern drug discovery workflows.
Fundamental Concepts of Protein-Ligand Interactions
The Nature of Non-Covalent Interactions
Non-covalent interactions form the physical basis of molecular recognition in biological systems, including drug-receptor binding. While traditional drug design has focused on well-known interactions like hydrophobic contacts, hydrogen bonds, and salt bridges, recent research highlights the significance of underappreciated interactions that substantially stabilize protein-ligand complexes [35]. These include:
- Cation-π interactions between electron-deficient aromatic systems and positively charged ions
- Halogen bonds where electropositive regions of halogens interact with electron donors
- CH-π and orthogonal multipolar interactions that contribute meaningfully to binding affinity
Understanding the complete repertoire of these interactions provides a more comprehensive framework for rational drug design and optimization.
Thermodynamic Principles of Binding
The binding affinity between a protein and ligand is governed by the Gibbs free energy change (ΔG) according to the relationship: ΔG = -RT ln(KA), where KA is the association constant (the reciprocal of KD). This free energy change results from the delicate balance between enthalpic (ΔH) and entropic (ΔS) contributions through the relationship ΔG = ΔH - TΔS [36].
Enthalpy-entropy compensation represents a fundamental challenge in rational drug design, where favorable enthalpic contributions (such as hydrogen bonding) often come at the cost of entropic penalties due to reduced flexibility in the bound state [36]. Additionally, the reorganization of water molecules around the binding site significantly influences the thermodynamics of binding, with approximately 20% of protein-bound waters being undetectable by X-ray crystallography [36].
Experimental Methods for KD Determination
Multiple biophysical techniques are available for characterizing protein-ligand interactions and determining dissociation constants. Each method offers distinct advantages, limitations, and appropriate application ranges, as summarized in Table 1.
Table 1: Comparison of Major Techniques for Protein-Ligand Interaction Analysis and KD Determination
| Technique | Principle | KD Range | Sample Consumption | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| ESI-MS | Mass-to-charge ratio measurement of intact complexes | Low nM to mM [34] | Very low (pmol amounts) [33] | Label-free, stoichiometry information, works with mixtures | Gas-phase vs. solution behavior may differ |
| Isothermal Titration Calorimetry (ITC) | Direct measurement of heat changes during binding | Low μM to nM [37] | Moderate to high | Provides full thermodynamic profile (KD, ΔH, ΔS, n) | Requires careful concentration determination [37] |
| Surface Plasmon Resonance (SPR) | Detection of refractive index changes near biosensor surface | High pM to low μM | Low | High throughput, real-time kinetics | Immobilization may alter protein behavior [32] |
| Microfluidic Diffusional Sizing (MDS) | Size change measurement via diffusion rates | Not specified | Low | Works in biological fluids, provides size information | Limited to fluorescently labeled probes [32] |
| NMR Spectroscopy | Chemical shift perturbations upon binding | mM to nM [36] | Moderate to high | Atomic-resolution, identifies binding sites, solution-state | Lower sensitivity, limited for large proteins |
Solution-Based versus Surface-Based Methods
A critical consideration in KD determination is the distinction between solution-based and surface-based methodologies. Surface-based techniques like SPR, BLI, and ELISA require immobilization of one binding partner on a surface, which can alter native conformation, introduce steric hindrance, and potentially affect activity [32]. Solution-based methods such as ESI-MS, ITC, and MDS maintain both interaction partners in their natural state, potentially providing more physiologically relevant measurements [32] [34].
ESI-MS Methodologies for KD Determination
Fundamental Principles of ESI-MS for Non-Covalent Complexes
Electrospray ionization mass spectrometry has emerged as a powerful technology for studying non-covalent ligand-macromolecular target interactions. The gentle nature of the ESI process allows for the transfer of non-covalent complexes from solution to the gas phase with preservation of binding stoichiometry and information about binding specificity [33]. Recent research into the mechanisms of ionization in charged microdroplets suggests that field ionization followed by chemical ionization occurs in water-containing microdroplets, generating reactive intermediates that may account for unique chemistry observed in these systems [18].
Experimental Workflows in ESI-MS
A typical ESI-MS workflow for KD determination involves incubating a constant concentration of protein with varying concentrations of ligand, directly introducing the mixture into the mass spectrometer via electrospray ionization, detecting and quantifying the free protein and protein-ligand complex signals in the mass spectrum, and calculating the KD from the concentration dependence of complex formation [38] [34].
For protein-ligand complexes prone to gas-phase dissociation, the reference ligand method provides enhanced accuracy. This approach involves adding a reference ligand (Lref) with known affinity that binds at the same site as the ligand of interest (L) and forms a stable complex in the gas phase. The fraction of protein bound to Lref, determined directly from the ES mass spectrum, is sensitive to the fraction bound to L in solution, enabling accurate determination of the affinity for L [38].
Diagram 1: ESI-MS Workflow for KD Determination. This diagram illustrates the key steps in determining dissociation constants using electrospray ionization mass spectrometry.
Practical Applications in Drug Discovery
ESI-MS has been successfully applied to various stages of drug discovery campaigns. In fragment-based drug discovery (FBDD) against targets like XIAP and CDK2, native ESI-MS derived KD values for compounds spanning low nanomolar to millimolar ranges, demonstrating excellent correlation with ITC-derived values [34]. The technology enables screening of compound libraries against multiple targets simultaneously (multitarget affinity/specificity screening), determining binding specificity, relative dissociation constants, and stoichiometry in a single assay [33].
Nanospray ionization, which uses smaller-diameter ESI emitters, offers several advantages for drug discovery applications, including significantly reduced sample consumption (meaningful measurements from 1-3 μL total volume) and potentially gentler ionization that better preserves non-covalent complexes [33].
Complementary and Orthogonal Techniques
Isothermal Titration Calorimetry (ITC)
ITC remains the gold standard for complete thermodynamic characterization of binding interactions, providing simultaneous determination of KD, enthalpy change (ΔH°), binding stoichiometry (n), and thereby the entropic contribution (ΔS) [37]. Recent developments include browser-based tools like ACI-ITC that calculate accuracy confidence intervals for ITC-derived parameters, explicitly accounting for systematic errors in reactant concentrations that can skew results [37].
NMR Spectroscopy
NMR spectroscopy provides atomic-resolution information about protein-ligand interactions in solution, complementing the structural information obtained from X-ray crystallography. NMR-driven structure-based drug design (NMR-SBDD) combines 13C side chain protein labeling strategies with computational tools to generate reliable protein-ligand structural ensembles that closely resemble native state distributions in solution [36]. NMR is particularly valuable for identifying hydrogen bonding interactions, as 1H chemical shifts directly report on the nature of hydrogen bonding, with downfield shifts indicating classical H-bonds and upfield shifts corresponding to CH-π interactions [36].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Materials for Studying Protein-Ligand Interactions
| Reagent/Material | Function and Importance | Application Examples |
|---|---|---|
| Stable Isotope-labeled Proteins (13C, 15N) | Enables NMR signal assignment; simplifies complex spectra | NMR-SBDD with selective side-chain labeling [36] |
| Reference Ligands | Binds specifically with known affinity; enables quantification of labile complexes | ESI-MS reference ligand method for complexes prone to gas-phase dissociation [38] |
| Biologically Relevant Buffers | Mimic physiological conditions; critical for physiologically relevant KD values | Testing affinities in serum, cell media rather than just aqueous buffers [32] |
| Cryo-EM Reagents | Preserve native protein structures for electron microscopy | Structural studies of large protein complexes inaccessible to X-ray crystallography [36] |
| Fragment Libraries | Collections of low molecular weight compounds for screening initial hits | Fragment-based drug discovery campaigns against protein targets like XIAP and CDK2 [34] |
Methodological Considerations and Best Practices
Avoiding Common Pitfalls in KD Determination
Several common mistakes can compromise the accuracy and interpretation of KD measurements:
- Ignoring experimental conditions: KD values are highly dependent on buffer composition, pH, ionic strength, and temperature. Always report detailed buffer conditions and standardize protocols during drug screening [32].
- Inappropriate binding models: Applying simple 1:1 binding models to systems with multiple binding sites or cooperative binding can yield incorrect KD values. Use sizing information or other orthogonal data to validate binding stoichiometry [32].
- Overreliance on single techniques: Corroborate ESI-MS findings with complementary methods like ITC or solution-based techniques to ensure results reflect solution-phase behavior rather than gas-phase artifacts [32] [34].
Data Interpretation and Validation
When applying ESI-MS to KD determination, consider that gas-phase stability does not always correlate directly with solution-phase affinity, as electrostatic and hydrogen-bonding interactions are strengthened in the gas phase while hydrophobic interactions are weakened [33]. The use of internal standards and reference ligands helps correct for these effects and instrument variability [38].
For ITC measurements, tools like ACI-ITC provide accuracy confidence intervals that account for systematic errors in concentration determinations, offering probabilistic ranges for true KD values and supporting more robust conclusions [37].
Accurate determination of dissociation constants remains essential for informed decision-making in drug discovery. ESI-MS methodologies provide powerful tools for characterizing non-covalent protein-ligand interactions, especially when integrated with orthogonal techniques like ITC and NMR spectroscopy. As research continues to unveil the complexities of molecular recognition—including underappreciated non-covalent interactions and the challenges of enthalpy-entropy compensation—the combination of multiple biophysical approaches will be crucial for advancing structure-based drug design. The continuing development of ESI-MS technologies and their application within drug discovery pipelines promises to enhance our ability to rapidly identify and optimize therapeutic compounds with precisely tailored binding properties.
Electrospray Ionization (ESI) has emerged as a cornerstone technique in analytical chemistry, enabling the sensitive and robust analysis of non-volatile and thermally labile biomolecules that are not amenable to traditional ionization methods [2]. This soft ionization technique uses electrical energy to assist the transfer of ions from solution into the gaseous phase before they are subjected to mass spectrometric analysis, making it particularly valuable for clinical and research applications [2]. ESI's capability to generate multiple charge ions has effectively extended the mass range of analyzers to accommodate the kiloDalton to megaDalton molecular weights observed in proteins and their associated peptides [3]. Within the context of metabolomics and therapeutic drug monitoring (TDM), ESI provides the fundamental ionization mechanism that enables high-throughput quantitative analysis of complex biological samples, driving advancements in personalized medicine and clinical diagnostics.
The significance of ESI in clinical laboratories has grown substantially over the past decade, with ESI-mass spectrometry (ESI-MS) providing a sensitive, robust, and reliable tool for studying metabolites and drugs at femto-mole quantities in micro-litre sample volumes [2]. When coupled with high-performance liquid chromatography (HPLC) for molecular fractionation prior to mass spectrometric analysis, HPLC/ESI-MS has become a powerful technique capable of analyzing both small and large molecules of various polarities in complex biological matrices [2]. The additional separation capabilities of tandem mass spectrometry (MS/MS) have further simplified complicated sample purification procedures, enabling the development of streamlined workflows for rapid analysis and high sample throughput that are essential for both metabolomics and therapeutic drug monitoring applications.
Fundamental Principles of Electrospray Ionization
The Electrospray Ionization Process
The transfer of ionic species from solution into the gas phase by ESI involves three distinct stages that transform sample molecules into gas-phase ions suitable for mass analysis [2] [3]. In the first stage, a fine spray of charged droplets is generated when the sample solution is pushed through a capillary tip maintained at a high voltage (typically 2.5-6.0 kV) relative to the surrounding chamber [2]. This application of high voltage to the liquid produces a Taylor cone, from which a fine mist of highly charged droplets with the same polarity as the capillary voltage is generated [39]. The use of a nebulizing gas (such as nitrogen) shearing around the eluted sample solution enhances the stability of this process and allows for higher sample flow rates [2].
In the second stage, these charged droplets pass down a pressure and potential gradient toward the analyzer region of the mass spectrometer [2]. With the aid of elevated ESI-source temperature and/or a stream of nitrogen drying gas, the charged droplets undergo continuous solvent evaporation, leading to a progressive reduction in droplet size and a corresponding increase in surface charge density [2]. As the solvent evaporates, the charge intensity on the droplet surface gradually increases until the droplet reaches the Rayleigh limit, where the Coulomb repulsive force between charges becomes sufficient to counteract the surface tension [3].
The third and final stage involves ion ejection from the highly charged droplets [2]. When the electric field strength within the charged droplet reaches this critical point, it becomes kinetically and energetically feasible for ions at the droplet surface to be emitted into the gaseous phase [2]. Two primary models explain the mechanism of gas-phase ion generation: the Charged Residue Model (CRM), which suggests that the droplet undergoes repeated fission events until a single ion remains, and the Ion Evaporation Model (IEM), which proposes that ions are directly emitted from the highly curved droplet surfaces before the droplet reaches the Rayleigh limit [3]. The emitted ions are then sampled by a skimmer cone and accelerated into the mass analyzer for subsequent analysis of molecular mass and measurement of ion intensity.
ESI Mass Spectrometry Configurations and Instrumentation
Multiple mass analyzer configurations can be coupled with ESI sources to address different analytical needs in metabolomics and therapeutic drug monitoring. The most common systems used in clinical and research settings include quadrupole, tandem quadrupole (triple-quad), ion trap, and high-resolution systems such as quadrupole time-of-flight (QqTOF) and Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometers [2] [40].
Quadrupole mass analyzers represent the workhorse instrumentation in clinical laboratories due to their robustness, economical operation, physically compact size, and compatibility with a wide variety of inlet systems [2]. These systems employ an assembly of four parallel metal rods maintained at equal distance, with opposite rods connected electrically [2]. A direct current (DC) voltage superimposed with a radio frequency (RF) alternating current voltage is applied to diagonally placed rod pairs, creating an oscillatory electrical field that filters ions based on their mass-to-charge (m/z) ratio [2].
Tandem quadrupole systems (triple-quad) incorporate three quadrupoles arranged linearly, enabling sophisticated fragmentation experiments essential for compound identification and quantification [2]. In this configuration, the first quadrupole (Q1) mass-selects a specific precursor ion, the second quadrupole (Q2) serves as a collision cell where collision-induced dissociation (CID) occurs using argon gas, and the third quadrupole (Q3) analyzes the resulting product ions [2]. This arrangement supports multiple data acquisition modes critical for quantitative analysis, including product ion scanning, precursor ion scanning, neutral loss scanning, and multiple reaction monitoring (MRM) [2]. The MRM mode, where both Q1 and Q3 are static for a predetermined pair of precursor and product ions, provides the highest specificity and sensitivity and is commonly used in ESI-MS/MS quantification procedures for both metabolomics and therapeutic drug monitoring [2].
Ion trap mass analyzers offer advantages in structural elucidation through multiple stages of fragmentation (MS^n capability) [2]. These systems consist of three hyperbolic electrodes that form a cavity for trapping and analyzing ions: a ring electrode positioned between two end cap electrodes [2]. Various voltages applied to these electrodes trap ions in stable oscillating trajectories confined within the trapping cell, with alterations to the electrode system potentials causing instabilities that eject ions in order of increasing m/z ratio for detection [2].
For applications requiring high mass accuracy and resolution, QqTOF and FT-ICR mass spectrometers provide superior performance [40]. The QqTOF configuration combines two quadrupoles with a time-of-flight analyzer, offering mass resolving power of approximately 10,000—sufficient for general mass distribution determination within samples [40]. In contrast, FT-ICR MS utilizes an ICR cell within a high magnetic field to achieve exceptional mass resolving powers exceeding 100,000 routinely, enabling the determination of exact molecular formulas for complex mixtures encountered in metabolomic studies [40].
High-Throughput Quantitative Analysis in Metabolomics
Advanced Methodologies for High-Throughput Metabolomics
Recent technological advancements have dramatically increased the throughput and sensitivity of metabolomic analyses, enabling researchers to process hundreds to thousands of samples with robust quantitative data. Emerging UPLC-MS methods now offer high-throughput MS analysis including chromatographic separation, allowing hundreds of MS-derived metabolomes to be acquired and analyzed in a single day [41]. These advancements in rapid acquisition approaches have facilitated high-throughput metabolomics-based discovery across diverse applications from clinical diagnostics to natural product identification [41].
A particularly innovative approach for single-cell metabolomics, termed HT SpaceM, demonstrates the cutting edge of high-throughput capability [42]. This method combines cell preparation on custom glass slides with small-molecule matrix-assisted laser desorption ionization (MALDI) imaging mass spectrometry and batch processing, enabling the analysis of over 140,000 cells from 132 samples with high reproducibility and single-cell resolution [42]. HT SpaceM can detect more than 100 small-molecule metabolites per cell from 500 to 1,000 cells per sample, identifying metabolic heterogeneity and pathway coordination at single-cell resolution that would be obscured in population-averaged measurements [42].
Another advanced platform for high-throughput single-cell metabolomics employs a serpentine channel microfluidic device coupled with pulsed electric field-induced electrospray ionization-high resolution mass spectrometry (PEF-ESI-HRMS) [43]. This system achieves continuous cell separation and inertial focusing, enabling single-cell analysis under near-physiological conditions at a throughput of up to 80 cells per minute [43]. Researchers utilizing this approach have detected more than 900 features and identified approximately 120 metabolites from a single cell, allowing discrimination of different cancer cell types based on their individual metabolic profiles through principal component analysis [43].
Experimental Protocol: High-Throughput Metabolomic Profiling Using LC-ESI-MS/MS
The following protocol outlines a standardized workflow for high-throughput metabolomic analysis using liquid chromatography coupled with ESI-tandem mass spectrometry:
Sample Preparation:
- Metabolite Extraction: Add 800 μL of cold methanol:water (80:20, v/v) to 200 μL of biological sample (plasma, serum, or cell culture extract) in a 2 mL microcentrifuge tube.
- Vortex and incubate: Mix vigorously for 30 seconds, then incubate at -20°C for 1 hour to precipitate proteins.
- Centrifugation: Centrifuge at 14,000 × g for 15 minutes at 4°C to pellet insoluble material.
- Supernatant collection: Transfer 750 μL of supernatant to a new LC-MS vial and evaporate to dryness under a gentle nitrogen stream.
- Reconstitution: Reconstitute the dried extract in 100 μL of LC-MS grade water:acetonitrile (95:5, v/v) containing internal standards appropriate for retention time alignment and signal normalization.
LC-ESI-MS/MS Analysis:
- Chromatographic separation: Employ a reversed-phase C18 column (100 × 2.1 mm, 1.8 μm) maintained at 40°C with a flow rate of 0.4 mL/min.
- Mobile phase: Utilize solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid) with the following gradient program: 0-2 min (2% B), 2-15 min (2-95% B), 15-18 min (95% B), 18-18.1 min (95-2% B), and 18.1-22 min (2% B) for column re-equilibration.
- ESI source parameters: Set capillary voltage to 3.5 kV (positive mode) or 3.0 kV (negative mode), source temperature to 150°C, desolvation temperature to 500°C, cone gas flow to 50 L/h, and desolvation gas flow to 800 L/h (nitrogen).
- Data acquisition: Operate the mass spectrometer in MRM mode with dwell times of 10-20 ms per transition, monitoring 100-300 metabolite-specific transitions per sample depending on the panel scope.
Data Processing and Analysis:
- Peak integration: Automatically integrate chromatographic peaks using vendor software with manual verification to ensure accuracy.
- Quality control: Apply quality assessment based on internal standard performance, with %CV < 15% for technical replicates and signal intensity drift < 20% across the batch.
- Statistical analysis: Perform multivariate statistical analysis including principal component analysis (PCA) and partial least squares-discriminant analysis (PLS-DA) to identify differentially abundant metabolites.
- Pathway analysis: Utilize metabolite set enrichment analysis and pathway mapping tools to identify biologically relevant metabolic pathways altered between experimental conditions.
Performance Metrics in High-Throughput Metabolomics
Table 1: Quantitative Performance Metrics of High-Throughput Metabolomics Platforms
| Platform/Technique | Throughput (Samples/Day) | Metabolites Detected | Sensitivity | Reproducibility (%CV) | Key Applications |
|---|---|---|---|---|---|
| Conventional LC-ESI-MS/MS | 50-100 | 100-300 targeted compounds | Low femtomole | 5-15% | Targeted metabolomics, clinical diagnostics |
| HT SpaceM (MALDI-MS) | 40 samples/slide | 100+ small molecules/cell | Not specified | <15% | Single-cell metabolomics, spatial mapping |
| Microfluidic PEF-ESI-HRMS | 80 cells/minute | 900+ features/cell | Not specified | Low variability | Cellular heterogeneity, cancer cell discrimination |
| UPLC-ESI-QTOF | 200-300 | 500-1000 untargeted features | High femtomole | 5-20% | Untargeted metabolomics, biomarker discovery |
| NanoESI-FT-ICR | 20-50 | >5000 features | Attomole-femtomole | 2-10% | Complex mixture analysis, natural products |
High-Throughput Approaches in Therapeutic Drug Monitoring
ESI-MS in Clinical TDM Applications
Electrospray ionization mass spectrometry has revolutionized therapeutic drug monitoring by enabling the simultaneous quantification of multiple drugs and their metabolites with exceptional sensitivity and specificity [2]. The capacity of ESI-tandem MS to measure biomolecules sharing similar molecular structures makes it particularly valuable for TDM applications where drug compounds and their metabolic derivatives must be quantified in complex biological matrices [2]. ESI-MS facilitates high-throughput TDM through its compatibility with liquid chromatography separation and ability to perform rapid multiple reaction monitoring (MRM) transitions, allowing clinical laboratories to process hundreds of patient samples daily with rapid turnaround times essential for dose adjustment decisions.
The application of ESI-MS in TDM spans numerous therapeutic classes, including immunosuppressants (tacrolimus, cyclosporine, sirolimus), antiepileptics (levetiracetam, valproic acid, carbamazepine), antibiotics (vancomycin, aminoglycosides), antiretroviral drugs (protease inhibitors, non-nucleoside reverse transcriptase inhibitors), and chemotherapeutic agents (methotrexate, imatinib) [2]. For each application, ESI-MS assays provide superior analytical performance compared to traditional immunoassays, with enhanced specificity through the discrimination of structurally similar compounds, improved sensitivity for monitoring drugs at low concentrations, and the capacity for multiplexing to simultaneously quantify parent drugs and their pharmacologically active metabolites in a single analytical run.
Experimental Protocol: Multiplexed TDM Analysis Using ESI-MS/MS
The following protocol details a validated approach for the simultaneous quantification of multiple therapeutic drugs in human plasma using LC-ESI-MS/MS:
Sample Preparation:
- Aliquot samples: Transfer 100 μL of calibrators, quality controls, and patient plasma samples to a 96-well protein precipitation plate.
- Protein precipitation: Add 300 μL of internal standard working solution in acetonitrile:methanol (50:50, v/v) containing stable isotope-labeled analogs of each target analyte.
- Mix and precipitate: Seal the plate, mix vigorously for 5 minutes, then centrifuge at 4,000 × g for 10 minutes at 10°C.
- Supernatant transfer: Transfer 150 μL of supernatant to a clean 96-well collection plate containing 150 μL of water to reduce organic solvent content.
- Seal and analyze: Seal the collection plate and place in the LC-ESI-MS/MS system autosampler maintained at 10°C.
LC-ESI-MS/MS Analysis:
- Chromatographic system: Utilize ultra-performance liquid chromatography with an ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 μm) maintained at 50°C.
- Mobile phase: Employ solvent A (2 mM ammonium acetate in water with 0.1% formic acid) and solvent B (2 mM ammonium acetate in methanol with 0.1% formic acid) with the following gradient: 0-0.5 min (10% B), 0.5-3.0 min (10-90% B), 3.0-4.0 min (90% B), 4.0-4.1 min (90-10% B), and 4.1-5.5 min (10% B) for re-equilibration at a flow rate of 0.5 mL/min.
- ESI source conditions: Set electrospray ionization in positive mode with capillary voltage 3.2 kV, source temperature 120°C, desolvation temperature 450°C, cone gas flow 50 L/h, and desolvation gas flow 800 L/h (nitrogen).
- MRM acquisition: Program the tandem mass spectrometer to monitor two MRM transitions per analyte (one quantifier, one qualifier) with optimized collision energies for each compound. Typical dwell times of 10-25 ms per transition enable adequate data point acquisition across chromatographic peaks.
Quantification and Quality Assurance:
- Calibration curve: Generate linear weighted (1/x²) calibration curves for each analyte using peak area ratios of analyte to internal standard.
- Quality control: Include at least three levels of quality control samples (low, medium, high) in each analytical batch with acceptance criteria of ±15% deviation from nominal concentrations.
- Carryover assessment: Monitor injection sequence for carryover by analyzing blank samples after high-concentration calibrators.
- Ion suppression/enhancement: Evaluate matrix effects using post-column infusion and post-extraction addition methods during method validation.
Analytical Validation Parameters for TDM Assays
Table 2: Validation Parameters for ESI-MS/MS-Based Therapeutic Drug Monitoring Assays
| Validation Parameter | Acceptance Criteria | Typical Performance | Importance in TDM |
|---|---|---|---|
| Accuracy | 85-115% of nominal value | 90-110% | Ensures correct concentration reporting for clinical decisions |
| Precision | Intra-day & inter-day CV < 15% | CV < 10% | Provides reproducible results across batches |
| Linearity | R² > 0.99 | R² > 0.995 | Enables quantification across therapeutic range |
| Lower Limit of Quantification | Signal-to-noise > 10, CV < 20% | 0.1-5 ng/mL | Determines lowest measurable concentration |
| Matrix Effects | Internal standard normalized matrix factor 85-115% | 90-110% | Accounts for ionization suppression/enhancement |
| Carryover | <20% of LLOQ in blank after high standard | Typically <1% | Prevents false elevation in subsequent samples |
| Stability | 85-115% of fresh sample | Typically meets criteria | Ensures reliability under storage conditions |
Essential Research Reagents and Materials
Table 3: Essential Research Reagents for ESI-MS Based Metabolomics and TDM
| Reagent/Material | Function | Application Examples | Technical Considerations |
|---|---|---|---|
| LC-MS Grade Solvents (water, methanol, acetonitrile) | Mobile phase components; sample preparation | Chromatographic separation; protein precipitation | Low UV absorbance; minimal volatile impurities |
| Volatile Buffers (ammonium acetate, ammonium formate) | Mobile phase additives | Improve ionization efficiency; control pH | Concentration typically 2-10 mM; compatible with ESI |
| Ion-Pairing Reagents (formic acid, acetic acid) | Enhance ionization in positive mode; TFA (caution) | Most small molecule applications in positive mode | Typically 0.05-0.1%; TFA can cause ion suppression |
| Stable Isotope-Labeled Internal Standards | Normalization for matrix effects; quantification correction | Deuterated drug analogs; ¹³C-labeled metabolites | Ideally ³-6 Da mass difference; identical chromatographic behavior |
| Solid Phase Extraction Cartridges | Sample clean-up; analyte enrichment | Phospholipid removal from plasma samples; concentration | Various chemistries (C18, mixed-mode, HILIC) for selectivity |
| Protein Precipitation Plates | High-throughput sample preparation | 96-well format for batch processing | Compatible with automation; minimal analyte adsorption |
| Quality Control Materials | Method validation; batch acceptance | Pooled human plasma; certified reference materials | Should mimic patient samples; multiple concentration levels |
Future Perspectives and Concluding Remarks
The field of high-throughput quantitative analysis in metabolomics and therapeutic drug monitoring continues to evolve rapidly, driven by technological advancements in electrospray ionization and mass spectrometry instrumentation. The electrospray ion source market is projected to grow at a compound annual growth rate of 6.3% from 2025 to 2032, reflecting increasing adoption across pharmaceutical, biotechnology, and clinical diagnostics sectors [44]. Key innovation trends focus on advanced miniaturization, enhanced sensitivity, integration of artificial intelligence for data processing, and development of multi-modal analysis systems that combine electrospray with complementary ionization techniques [44].
Future developments in ESI technology will likely focus on improving quantitative performance while further increasing analytical throughput. The emergence of ambient ionization techniques that require minimal sample preparation, coupled with advances in mass spectrometer miniaturization for point-of-care testing, promise to expand the applications of ESI-MS in routine clinical practice [45]. Additionally, the integration of artificial intelligence and machine learning algorithms for automated data interpretation and quality control will enhance the reproducibility and reliability of high-throughput analyses [44]. These technological advancements, combined with the established strengths of ESI in analyzing complex biological mixtures, ensure that electrospray ionization will remain a foundational technology in clinical and research mass spectrometry, continuing to enable new discoveries in metabolomics and improved patient care through precision therapeutic drug monitoring.
Maximizing Performance: A Strategic Guide to ESI Source Optimization and Problem-Solving
Electrospray Ionization (ESI) has profoundly transformed mass spectrometry, enabling the analysis of large, non-volatile, and thermally labile biomolecules directly from liquid solutions [2] [46]. The technique serves as a critical interface between liquid-phase separation methods like HPLC and the gas-phase analysis environment of the mass spectrometer, making it indispensable in modern drug development and proteomic research [2] [47]. The central thesis of this work posits that robust and reproducible ESI-MS data are contingent upon a foundational understanding of the ionization mechanism, coupled with the systematic optimization of three core instrumental parameters: sprayer voltage, and gas flow rates and temperature. These parameters collectively govern the efficiency of droplet formation, desolvation, and ultimate ion liberation, directly impacting method sensitivity, stability, and quantitative accuracy [48] [2]. This guide provides an in-depth examination of these critical adjustments, contextualized within the framework of how electrospray ionization functions, to empower researchers in achieving optimal analytical performance.
Theoretical Foundation of Electrospray Ionization
The electrospray process facilitates the transfer of ions from solution into the gas phase through a series of orchestrated events. A fundamental comprehension of this mechanism is vital for rational parameter optimization rather than reliance on empirical tuning alone.
The ESI Mechanism: From Liquid to Gas Phase
The transfer of ionic species from solution into the gas phase by ESI involves three primary steps, as depicted in Figure 1 [2]:
- Droplet Formation and Charging: A high voltage (typically 2.5–6.0 kV) is applied to a capillary through which the liquid sample flows. This induces a mist of highly charged droplets at the capillary tip, with the same polarity as the applied voltage [2] [49]. The application of a nebulizing gas shears the liquid stream, enhancing droplet formation and supporting higher flow rates [2].
- Solvent Evaporation and Droplet Shrinking: The charged droplets travel toward the mass spectrometer inlet under atmospheric pressure. With the aid of a heated drying gas, solvent evaporates from the droplet surface, causing the droplets to shrink. This reduction in size increases the surface charge density until it reaches the Rayleigh stability limit [2] [46].
- Ion Ejection: Upon reaching this critical charge density, the droplet undergoes Coulombic fission, disintegrating into smaller, progeny droplets. Two primary models explain the final stage of ion release:
- Charge Residue Model (CRM): Proposes that successive solvent evaporation and droplet fission cycles continue until a droplet containing a single analyte molecule remains. The solvent finally evaporates, leaving the analyte with the residual charge [46].
- Ion Evaporation Model (IEM): Suggests that when the droplets become sufficiently small (∼20 nm), the electric field at the surface is strong enough to directly desorb or "evaporate" solvated ions into the gas phase [46].
In practice, both mechanisms likely contribute to the final ion population detected, with their relative dominance depending on the analyte and experimental conditions [46].
Figure 1. Electrospray Ionization Mechanism Workflow. This diagram illustrates the sequential process from charged droplet formation to the generation of gas-phase ions via two predominant theoretical models.
Critical Parameter Adjustment
The stability and intensity of the ESI signal are highly dependent on the interplay of several source parameters. Mastering the adjustment of sprayer voltage, gas flows, and temperature is paramount for method development.
Sprayer Voltage Optimization
The voltage applied to the ESI capillary (sprayer voltage) is the primary driver for initiating and sustaining the electrospray. Its optimal setting is a balance between achieving sufficient ionization and avoiding disruptive electrical phenomena.
- Function and Typical Range: The sprayer voltage (typically 2–6 kV) creates the strong electric field necessary to disperse the liquid sample into a fine mist of charged droplets and guides them toward the counter-electrode [2] [49].
- Optimization Strategy: While often set to a default value, fine-tuning the voltage for specific analytes or mobile phase compositions can yield significant sensitivity gains. A systematic approach involves infusing the analyte and incrementally adjusting the voltage while monitoring the signal intensity of the precursor ion. The goal is to find the voltage that provides a stable, maximum signal.
- Critical Considerations:
- Electrical Discharge: In negative ion mode, or with highly aqueous mobile phases, excessively high voltages can cause electrical discharge (corona discharge), leading to signal instability and complete loss of signal. This can be identified by the appearance of protonated solvent clusters (e.g., H₃O⁺(H₂O)ₙ) in positive mode [48].
- Rim Emission: At very high voltages, the electrospray can become unstable, emitting not from the tip of the Taylor cone but from its rim, resulting in an erratic signal [48].
- Mobile Phase Composition: The optimal voltage is influenced by the surface tension of the mobile phase. More aqueous solvents have higher surface tension and require higher sprayer potentials. Adding a small amount of a organic modifier like methanol or isopropanol (1–2% v/v) can lower surface tension, allowing for stable spraying at lower voltages and often improving response [48].
Table 1: Guidelines for Sprayer Voltage Optimization
| Parameter | Typical Range | Optimization Goal | Common Pitfalls |
|---|---|---|---|
| Sprayer Voltage | 2.0 – 6.0 kV [2] [49] | Maximize precursor ion signal stability and intensity. | Electrical discharge (esp. in negative mode), rim emission, unwanted redox reactions [48]. |
| Voltage & Solvent | Adjust based on % aqueous: High for aqueous, Low for organic | Stable Taylor cone formation with lowest possible current. | High aqueous content requires higher voltage but increases discharge risk [48]. |
| Additive Use | 1-2% v/v Methanol or IPA | Lower surface tension to enable stable spraying at lower voltage. | May slightly alter chromatography; use consistent grade [48]. |
Gas Flow Rates and Temperature Optimization
The nebulizing, desolvation, and drying gases, along with their associated temperatures, are critical for controlling the droplet size and the efficiency of solvent evaporation throughout the ESI process.
- Nebulizer Gas: This is typically nitrogen that flows concentrically around the capillary tip. Its primary function is to shear the liquid stream into smaller, more uniform primary droplets, which facilitates easier desolvation. The optimal flow rate is dependent on the LC flow rate; it must be tuned to produce a stable spray without causing turbulence that can deflect ions away from the inlet [48] [47].
- Drying/Desolvation Gas: This stream of heated nitrogen flows across the path of the charged droplets. Its purpose is to accelerate the evaporation of solvent from the droplets, driving the process of droplet shrinkage and subsequent ion release. The temperature must be high enough to ensure complete desolvation (preventing solvent clusters from entering the mass analyzer) but not so high as to thermally degrade the analyte or disrupt non-covalent complexes [48] [2].
- Optimization Strategy: These parameters are highly inter-related. A common strategy is to use a design of experiments (DoE) approach to efficiently find the optimal combination. For initial setup, begin with manufacturer-recommended settings (e.g., nebulizer gas: 10-50 psi, drying gas: 4-12 L/min, temperature: 200-340 °C) and adjust while monitoring the signal of a known standard [48] [47].
Table 2: Optimization of Gas Flow and Temperature Parameters
| Parameter | Primary Function | Typical Range | Impact of High Setting | Impact of Low Setting |
|---|---|---|---|---|
| Nebulizer Gas Pressure | Shear liquid into fine primary droplets; stabilize spray [48]. | 10 - 50 psi [47] | Turbulence, ion deflection, signal loss. | Large droplet size, poor desolvation, unstable spray. |
| Drying Gas Flow Rate | Remove solvent vapor; assist droplet desolvation [48] [2]. | 4 - 12 L/min [47] | Premission of droplets/ions, potential signal decrease. | Incomplete desolvation, high chemical noise, solvent clusters. |
| Drying Gas Temperature | Provide energy for rapid solvent evaporation [48] [49]. | 200 - 340 °C [47] [49] | Thermal degradation of labile analytes or complexes. | Incomplete desolvation, reduced sensitivity, high background. |
The relationships between these core parameters and the resulting signal quality are complex, as summarized in Figure 2.
Figure 2. Parameter Interrelationships and Signal Outcome. This diagram visualizes how individual ESI source parameters affect physical processes and the potential pitfalls of improper settings, all converging on the final signal quality.
Advanced Methodologies: Systematic Optimization
For demanding applications such as the quantification of metabolites or the study of non-covalent protein-ligand complexes, a more rigorous approach to parameter optimization is required beyond univariate (one-variable-at-a-time) tuning.
Design of Experiments (DoE) for ESI Optimization
The Design of Experiments (DoE) is a powerful multivariate statistical approach that allows for the efficient evaluation of multiple factors and their interactions on a response (e.g., signal intensity) in a minimal number of experimental runs [50] [47]. This method is superior to the one-variable-at-a-time approach because it can reveal interaction effects that would otherwise be missed.
- Screening Phase: The process often begins with a screening design, such as a Two-Level Fractional Factorial Design (FFD). This design is used to identify which factors (e.g., capillary voltage, nebulizer pressure, drying gas flow, and temperature) have a statistically significant effect on the MS response. An FFD investigates f factors in N = 2f-v experiments, making it highly efficient [47].
- Optimization Phase: Once the significant factors are identified, an optimization design like a Central Composite Design (CCD) or Box-Behnken Design (BBD) is employed. These designs model the response surface, allowing researchers to pinpoint the optimal parameter settings and understand the curvature of the response [50] [47].
- Application Example: In a study to optimize the determination of metabolites in human urine, a four-factor, two-level FFD was first used to screen capillary voltage, nebulizer pressure, gas flow rate, and gas temperature. The significant factors were then optimized using a face-centered CCD for positive ion mode and a BBD for negative ion mode, resulting in a significant increase in sensitivity for problematic analytes like 7-methylguanine and glucuronic acid [47].
Experimental Protocol: DoE for ESI Source Optimization
Objective: To systematically identify the optimal combination of ESI source parameters (Sprayer Voltage, Nebulizer Gas, Drying Gas Flow, and Drying Gas Temperature) to maximize the signal intensity for a target analyte.
Materials & Reagents:
- Mass spectrometer with an ESI source and controllable parameters.
- HPLC system or syringe pump for isocratic delivery.
- Standard solution of the target analyte (e.g., 0.05 - 1 µg/mL in a compatible solvent).
- ESI-compatible mobile phase (e.g., 0.06% acetic acid in water/acetonitrile).
Methodology:
- Define Factors and Ranges: Set the factors to be investigated and their practical ranges based on instrument limits and preliminary experiments (e.g., Capillary Voltage: 2000-4000 V; Nebulizer Pressure: 10-50 psi; Drying Gas Flow: 4-12 L/min; Drying Gas Temperature: 200-340 °C) [47].
- Create Experimental Design: Use statistical software (e.g., JMP, R) to generate a design matrix. For a screening phase, a Fractional Factorial Design is appropriate. For optimization, a Central Composite Design is recommended.
- Execute Experiments: Run the experiments in the order specified by the design matrix. The response variable is the peak area or height of the target analyte's ion (e.g., in MRM mode) [47].
- Statistical Analysis and Modeling: Fit the experimental data to a mathematical model (e.g., a quadratic polynomial) using Response Surface Methodology (RSM). The software will generate coefficients for each factor and their interactions.
- Interpretation and Validation: Analyze the model to identify significant factors and locate the optimum conditions. Generate response surface plots for visualization. Finally, validate the predicted optimum by running the instrument at the suggested settings and confirming that the signal response matches the prediction.
The Scientist's Toolkit: Essential Research Reagents and Materials
The quality of reagents and materials used in ESI-MS is critical for minimizing background interference and ensuring robust performance.
Table 3: Essential Research Reagents and Materials for ESI-MS
| Item | Function & Importance | Key Considerations |
|---|---|---|
| LC-MS Grade Solvents (Water, Acetonitrile, Methanol) | Dissolve samples and constitute mobile phase. High purity is essential to minimize background ions and suppress analyte signal. | Avoid use of stabilizers (e.g., plasticizers) or preservatives that can cause ion suppression and contaminate the source [48] [23]. |
| Volatile Additives (Formic Acid, Acetic Acid, Ammonium Acetate) | Adjust pH to promote analyte protonation/deprotonation and improve chromatographic peak shape. | Typically used at 0.1% (acids) or 1-10 mM (buffers). Phosphate buffers and ion-pairing reagents should be avoided as they suppress ionization and contaminate the system [23]. |
| High-Purity Nitrogen Gas | Serves as the nebulizing and drying gas. Critical for stable spray formation and efficient desolvation. | Standard laboratory supply is sufficient. Must be oil-free to prevent source contamination. |
| Plastic vs. Glass Vials | Sample containers. Plastic is generally preferred for aqueous samples to avoid leaching of metal ions from glass. | Metal ions (Na+, K+) from glass vials can form metal adducts [M+Na]+, complicating spectra and reducing [M+H]+ signal. Plasticizers from certain plastics can also leach, but are typically less problematic [48]. |
| Sample Preparation Consumables (SPE cartridges, filters) | Purify and remove salts and matrix interferences from complex samples (e.g., biological fluids). | High salt content causes ion suppression, adduct formation, and poor quantitative results. Rigorous sample clean-up is key for analyzing biological matrices [48] [2]. |
The precision adjustment of sprayer voltage, gas flow rates, and temperature is not a mere procedural step but a fundamental scientific undertaking grounded in the principles of electrospray ionization. As detailed in this guide, these parameters directly control the physical processes that transform a liquid sample into a usable gas-phase ion beam. Moving beyond rudimentary tuning to embrace systematic methodologies like Design of Experiments empowers researchers to unlock the full potential of their LC-MS systems. This rigorous approach is indispensable for tackling the complex analytical challenges in modern drug development, from quantifying low-abundance metabolites in biological matrices to characterizing the delicate interactions of non-covalent protein-ligand complexes. By mastering these critical parameter adjustments, scientists can ensure their ESI-MS methods are not only sensitive and robust but also fundamentally sound.
Systematic Optimization Using Design of Experiments (DOE) and Response Surface Methodology (RSM)
Electrospray Ionization (ESI) is a soft ionization technique that has profoundly transformed biological and chemical analysis by enabling the transfer of ions from solution to the gas phase for mass spectrometric analysis [51]. As the most widely used ionization technique in chemical and biochemical analysis today, ESI allows the investigation of molecular composition of liquid samples, accommodating everything from small metabolites to large noncovalent protein complexes without mass limitations [51]. The ESI process involves three fundamental steps: dispersal of a fine spray of charged droplets, solvent evaporation, and ion ejection from the highly charged droplets into the gas phase [2]. However, the efficiency of this process depends critically on multiple interdependent parameters that must be carefully optimized to achieve maximum sensitivity and reliability.
The optimization of ESI conditions presents a significant challenge for researchers and drug development professionals. Traditional one-variable-at-a-time (OVAT) approaches, where each parameter is optimized individually while keeping others constant, are inefficient and often unsuccessful because they ignore interactions between factors and only explore a small part of the experimental domain [47]. In complex systems like ESI-mass spectrometry (ESI-MS), where numerous parameters can influence ionization efficiency, a systematic approach using Design of Experiments (DOE) and Response Surface Methodology (RSM) provides a more effective strategy for method development [50] [47] [52]. This technical guide explores the application of DOE and RSM for optimizing ESI conditions, providing detailed methodologies, experimental protocols, and practical implementation strategies for researchers seeking to enhance their analytical capabilities.
Theoretical Foundation: DOE and RSM Principles
Design of Experiments (DOE) Fundamentals
DOE represents a collection of statistical methods that enable researchers to efficiently plan and conduct experiments by systematically varying input factors to determine their effects on response variables [47]. Unlike OVAT approaches, DOE varies all relevant factors simultaneously according to a predefined experimental matrix, allowing for the identification of factor interactions while minimizing the number of required experimental runs [50]. The mathematical foundation of DOE typically involves building polynomial models to describe the relationship between factors and responses. For screening designs, first-order models are often employed:
[y = \beta0 + \sum{i=1}^{k}\betaixi + \varepsilon]
where (y) is the response, (\beta0) is the constant term, (\betai) represents the coefficient of the ith factor, (x_i) is the ith factor, and (\varepsilon) is the residual associated with the experiments [47].
For optimization studies, more complex second-order models including quadratic terms are typically used:
[y = \beta0 + \sum{i=1}^{k}\betaixi + \sum{i=1}^{k}\beta{ii}xi^2 + \sum{1≤i
This equation includes linear terms, quadratic terms, and interaction terms between factors, providing a comprehensive model of the response surface [52].
Response Surface Methodology (RSM) Framework
RSM encompasses mathematical and statistical techniques for building empirical models that describe how input factors influence one or more responses [53]. By fitting a polynomial equation to experimental data, RSM enables researchers to simultaneously optimize multiple factors to achieve optimal system performance [53]. The methodology typically involves a sequential approach beginning with screening experiments to identify significant factors, followed by response surface designs to model curvature and locate optimum conditions [52].
Several experimental designs are available for RSM, each with distinct advantages:
Central Composite Design (CCD): A popular design that combines a two-level factorial design with axial points and center points, allowing estimation of second-order effects [50]. CCD can be used in inscribed (CCI), face-centered, or circumscribed variations depending on factor constraints.
Box-Behnken Design (BBD): A three-level spherical design that combines two-level factorial arrangements with incomplete block designs [52]. BBD is often more efficient than CCD as it requires fewer experimental runs, especially when the number of factors is moderate [52].
Optimal Designs (OD): Computer-generated designs that optimize various statistical properties, including OD-A (minimizes average variance of coefficients), OD-D (minimizes confidence ellipsoid volume), OD-I (minimizes average prediction variance), OD-Distance, and OD-Modified Distance [53].
The choice among these designs depends on the specific experimental goals, number of factors to be optimized, and practical constraints such as available resources and time.
The Systematic Optimization Workflow for ESI-MS
The application of DOE and RSM to ESI optimization follows a logical sequence from screening to optimization and finally validation. This workflow ensures that all critical factors are identified and properly tuned to achieve optimal performance.
Figure 1: Systematic ESI Optimization Workflow. This diagram illustrates the sequential process for optimizing electrospray ionization parameters using DOE and RSM methodologies.
ESI Parameters for Systematic Optimization
Critical ESI Source Parameters
Multiple ESI source parameters significantly impact ionization efficiency and must be considered during method optimization. Based on experimental studies, the following parameters have been identified as most influential:
Gas and Temperature Parameters:
- Nebulizer Gas Pressure: Sheaths the liquid sample to facilitate droplet formation [2]. Typically ranges from 10-50 psi [47].
- Drying Gas Flow Rate: Aids in solvent evaporation from charged droplets [50]. Often optimized between 4-12 L/min [47].
- Drying Gas Temperature: Affects desolvation rate of charged droplets [50]. Commonly adjusted from 200-340°C [47].
Electrical Parameters:
- Capillary Voltage: Applied high voltage (2-6 kV) to create charged droplets [49]. Significantly impacts electrospray stability.
- Capillary Exit Voltage: Influces ion transfer efficiency into the mass analyzer [50].
- Skimmer Voltages: Multiple skimmer voltages (Skimmer 1, Skimmer 2) affect ion focusing and transmission [50].
Additional Source Parameters:
- Sample Flow Rate: Affects droplet size and ionization efficiency, with lower flow rates (nL/min) generally providing better sensitivity [1].
- Ion Transfer Tube Temperature: Optimized between 215-266°C to balance desolvation and analyte stability [53].
- Vaporizer Temperature: Affects solvent evaporation, typically optimized between 220-320°C [53].
Experimental Response Metrics
When optimizing ESI conditions, researchers typically monitor one or more of the following response metrics:
- Ion Abundance: Total signal intensity for the target analytes [50].
- Signal-to-Noise Ratio: Ratio of analyte signal to background noise [52].
- Relative Ionization Efficiency: Ratio of protein-ligand complex to free protein ion abundances (PL/P) in binding studies [50].
- Chromatographic Response: Peak area or height for LC-ESI-MS applications [52] [53].
- Sensitivity: Often measured as limit of detection (LOD) or limit of quantification (LOQ) [52].
Table 1: Critical ESI Parameters for Optimization in LC-MS Applications
| Parameter Category | Specific Parameters | Typical Range | Influence on ESI Process |
|---|---|---|---|
| Gas Settings | Nebulizer Gas Pressure | 10-50 psi [47] | Affects initial droplet formation and spray stability |
| Drying Gas Flow Rate | 4-12 L/min [47] | Influences solvent evaporation rate | |
| Drying Gas Temperature | 200-340°C [47] | Controls desolvation efficiency | |
| Voltage Settings | Capillary Voltage | 2000-4000 V [47] | Determines droplet charging and ESI stability |
| Capillary Exit Voltage | Instrument-specific | Affects ion transfer into mass analyzer | |
| Skimmer Voltages | Instrument-specific | Influences ion focusing and transmission | |
| Temperature Settings | Vaporizer Temperature | 220-320°C [53] | Impacts solvent evaporation |
| Ion Transfer Tube Temperature | 215-266°C [53] | Balances desolvation and analyte stability | |
| Flow Settings | Sample Flow Rate | nL/min to μL/min [1] | Affects droplet size and ionization efficiency |
Experimental Design Strategies for ESI Optimization
Screening Designs: Identifying Critical Factors
The first step in systematic ESI optimization involves screening a broad set of potential factors to identify those with significant effects on the response. Fractional Factorial Designs (FFD) are particularly valuable for this purpose as they allow researchers to examine many factors with a minimal number of experimental runs [47]. A FFD with two levels (high and low) for each factor follows the formula (N = 2^{f-v}), where (f) is the number of factors, (v) determines the fraction of the full factorial, and (N) is the required number of experiments [47].
For example, in optimizing ESI conditions for the determination of metabolites in human urine, researchers employed a four-factor, two-level FFD to screen capillary voltage, nebulizer pressure, gas flow rate, and gas temperature [47]. This approach efficiently identified the most influential factors before proceeding to more detailed optimization.
Response Surface Designs: Modeling and Optimization
Once critical factors are identified, response surface designs are employed to build predictive models and locate optimal conditions. Different design strategies offer distinct advantages:
Central Composite Design (CCD): CCD includes three types of points: factorial points from a (2^k) design, axial points positioned at ±α from the center, and center points to estimate pure error [50]. The value of α depends on desired design properties, with α = 1 used for face-centered designs where factors have strict upper and lower limits [47]. The total number of experiments required for CCD with (k) factors is (2^k + 2k + C), where (C) is the number of center points [50].
Box-Behnken Design (BBD): BBD is a three-level spherical design that doesn't contain combinations where all factors are at their extreme settings simultaneously [52]. This can be advantageous when extreme conditions may cause experimental difficulties. BBD requires fewer runs than CCD when the number of factors is small to moderate, making it efficient for ESI optimization [52].
Optimal Designs (OD): ODs are computer-generated based on specific optimality criteria and can be more flexible and efficient than traditional designs, especially when dealing with constraints or unusual experimental regions [53]. Different OD types optimize different statistical properties:
- OD-A: Minimizes average variance of regression coefficients
- OD-D: Maximizes determinant of the information matrix
- OD-I: Minimizes average prediction variance
- OD-Distance: Maximizes Euclidean distance between design points
- OD-Modified Distance: Augments design matrix rank [53]
Table 2: Comparison of Experimental Designs for ESI Optimization
| Design Type | Number of Experiments for 3 Factors | Key Features | Best Use Cases |
|---|---|---|---|
| Full Factorial | 8 (2-level) to 27 (3-level) | Estimates all main effects and interactions | Initial screening with limited factors |
| Fractional Factorial | 4-16 depending on resolution | Reduces run number while estimating main effects | Screening many factors efficiently |
| Central Composite (CCD) | 15-20 including center points | Estimates curvature with axial points | Comprehensive optimization with 2-6 factors |
| Box-Behnken (BBD) | 15 including center points | No extreme conditions; spherical design | Safe optimization avoiding factor extremes |
| Optimal Designs (OD) | Variable based on criteria | Computer-generated for specific optimality | Constrained regions or specific precision needs |
Case Study: Protein-Ligand Binding Studies
A comprehensive example of ESI optimization comes from protein-ligand binding studies between Plasmodium vivax guanylate kinase (PvGK) and its ligands GMP and GDP [50]. Researchers employed inscribed central composite designs (CCI) to optimize ESI conditions for accurate determination of equilibrium dissociation constants (K~D~). The experimental design included five factors studied at five levels each, with orthogonal and rotatable properties to ensure precise estimation of response surfaces [50].
The response metric was the relative abundance ratio of protein-ligand complex to free protein (PL/P), which needs to reflect solution-phase equilibrium concentrations accurately. Even for the structurally similar ligands GMP and GDP, different optimal ESI conditions were required, highlighting the importance of system-specific optimization [50].
Implementation Protocols and Methodologies
Step-by-Step Optimization Procedure
Phase 1: Pre-Optimization Preparation
- Define Optimization Goals: Clearly identify primary responses (e.g., signal intensity, complex stability, sensitivity) and any constraints (e.g., flow rate limitations, temperature sensitivities).
- Select Factors and Ranges: Based on preliminary knowledge and literature, choose factors to investigate and establish practical ranges that cover feasible operating conditions.
- Prepare Standards and Reagents: Use stable standard solutions at concentrations relevant to actual samples. For biological applications, use appropriate buffers such as 10 mM ammonium acetate (pH 6.8) to maintain native conditions [50].
Phase 2: Experimental Execution
- Generate Experimental Design: Use statistical software to create the design matrix, randomizing run order to minimize systematic bias.
- Conduct ESI-MS Analysis: Perform experiments according to the design matrix, ensuring consistent sample introduction and stable instrument performance.
- Record Response Data: Collect response measurements with sufficient replication to estimate experimental error.
Phase 3: Data Analysis and Optimization
- Build Response Models: Fit appropriate mathematical models (typically second-order polynomials) to the experimental data.
- Evaluate Model Adequacy: Check statistical measures (R², adjusted R², predicted R²) and examine residual plots to verify model validity.
- Locate Optimum Conditions: Use response surface plots and numerical optimization to identify factor settings that maximize desirability.
- Verify Predictions: Conduct confirmation experiments at predicted optimum conditions to validate model accuracy.
Advanced Optimization with Derringer's Desirability Function
For multi-response optimization, Derringer's desirability function provides a powerful approach to simultaneously optimize multiple criteria [52]. This method converts each response into an individual desirability function (ranging from 0 to 1) and then combines them into an overall composite desirability. The major advantage is that if one criterion is not met, the overall product becomes unacceptable, ensuring balanced optimization across all responses [52].
In the analysis of alkaloids in Meconopsis species, researchers applied Derringer's desirability to simultaneously optimize UHPLC separation and ESI sensitivity, achieving a nearly 10-fold improvement in detection limits compared to previous methods [52].
Research Reagent Solutions for ESI-MS Optimization
Successful ESI optimization requires appropriate selection of reagents and materials that facilitate efficient ionization while maintaining analytical integrity.
Table 3: Essential Research Reagents for ESI-MS Method Development
| Reagent Category | Specific Examples | Function in ESI-MS | Application Notes |
|---|---|---|---|
| Volatile Buffers | Ammonium acetate (10 mM) [50] | Maintains solution conductivity without ESI suppression | Compatible with native MS for protein complexes |
| LC-MS Solvents | Acetonitrile (LC-MS grade) [52] | Organic modifier for mobile phase | Enhorses droplet evaporation and ionization efficiency |
| Water (LC-MS grade) [52] | Aqueous component of mobile phase | Minimal impurities reduce background noise | |
| Ionization Additives | Formic acid (0.06%) [47] | Provides protons for positive ion mode ionization | Optimizes [M+H]+ formation for small molecules |
| Acetic acid [1] | Alternative proton source | milder acidity for sensitive compounds | |
| Calibration Standards | ESI-L Low Concentration Tuning Mix [47] | Instrument calibration and mass accuracy verification | Essential for method validation and transfer |
| Chemical Derivatization Reagents | FMOC (9-fluorenylmethyl chloroformate) [53] | Enhances detection sensitivity for amino acids | Critical for low-abundance analyte analysis |
Data Analysis and Interpretation Strategies
Response Surface Visualization and Interpretation
Response surface plots provide powerful visual tools for understanding factor effects and identifying optimum conditions. These three-dimensional representations show how a response variable changes as a function of two factors while holding other factors constant.
Figure 2: Response Surface Interpretation Logic. This diagram illustrates the relationship between experimental factors and response surface modeling for identifying optimum conditions.
When interpreting response surfaces, several characteristic shapes indicate different types of factor relationships:
- Linear Surfaces: Suggest a monotonic relationship between factors and response
- Curved Surfaces with Maximum: Indicate an optimum exists within the experimental region
- Saddle Shapes: Suggest complex interactions between factors requiring careful optimization
Statistical Validation of Models
Adequate model validation is essential before implementing optimization recommendations. Key statistical measures include:
- Model Significance: Evaluated through Analysis of Variance (ANOVA) with p-values < 0.05 indicating significant models.
- Lack-of-Fit Test: Distinguishes residual error from pure error, with non-significant lack-of-fit (p > 0.05) indicating good model fit.
- Coefficient of Determination (R²): Represents the proportion of variance explained by the model, with values > 0.80 generally acceptable.
- Predicted R²: Measures how well the model predicts new data, should be in reasonable agreement with adjusted R².
In the optimization of Gln-FMOC derivative analysis, researchers compared multiple DOE approaches and found that OD-A generated superior response compared to other designs, producing both high-performance experimental results and accurate forecasted surface plot maximal responses [53].
Applications in Pharmaceutical and Biomedical Research
The systematic optimization of ESI conditions using DOE and RSM has enabled significant advances in pharmaceutical and biomedical research:
Drug Discovery and Development:
- Protein-ligand binding constant determination under native conditions [50]
- High-throughput screening of compound libraries with enhanced sensitivity
- Metabolite identification and quantification in drug metabolism studies
Clinical Diagnostics:
- Sensitive detection of biomarkers in complex biological matrices [2]
- Newborn screening for inborn errors of metabolism [2]
- Hemoglobin variant analysis for hematological disorders [2]
Biomolecular Characterization:
- Stoichiometry determination of noncovalent protein complexes [51]
- Protein folding and conformational studies [50]
- Post-translational modification analysis
The application of systematic optimization approaches has been particularly valuable in quantitative bioanalysis, where robust and sensitive methods are required for regulatory submissions. The improved reproducibility and reliability of DOE-optimized methods facilitate method transfer between laboratories and instruments.
Systematic optimization using DOE and RSM represents a powerful paradigm for developing robust and sensitive ESI-MS methods. By simultaneously considering multiple factors and their interactions, researchers can efficiently navigate complex experimental spaces to identify optimal conditions that would be difficult to discover using traditional OVAT approaches. The structured methodology not only improves analytical performance but also provides deeper understanding of the ESI process and its dependence on various instrumental parameters.
As ESI-MS continues to evolve with new applications in proteomics, metabolomics, and pharmaceutical analysis, the implementation of systematic optimization strategies will remain essential for harnessing the full potential of this versatile ionization technique. The integration of advanced experimental designs with robust statistical analysis provides a framework for continuous method improvement that keeps pace with the increasing demands of modern analytical science.
Electrospray Ionization (ESI) is a cornerstone soft ionization technique in modern mass spectrometry, enabling the analysis of large, non-volatile biomolecules such as proteins and peptides by generating gas-phase ions directly from liquid solutions. [54] [1] Its mechanism involves applying a high voltage to a liquid, creating a Taylor cone and emitting a fine aerosol of charged droplets. Through solvent evaporation and repeated Coulomb fissions, these droplets eventually produce gas-phase analyte ions, often with multiple charges. [54] [1] This "soft" process preserves molecular integrity, minimizing fragmentation. [1] The transformative impact of ESI was recognized in 2002 with the Nobel Prize in Chemistry awarded to John B. Fenn. [54]
Despite its revolutionary role, the practical application of ESI is fraught with technical challenges that can compromise data quality. Salt adducts, ion suppression, and electrical discharge represent three prevalent issues that significantly impact the sensitivity, accuracy, and reproducibility of ESI-mass spectrometry (ESI-MS) analyses. Salt adducts, typically formed with species like sodium or potassium, lead to peak splitting and spectral complexity, obscuring the true molecular ion signal. [55] Ion suppression, a matrix effect, reduces ionization efficiency for target analytes in the presence of other compounds, thereby lowering sensitivity and hindering accurate quantification. [54] [56] Electrical discharge, an uncontrolled electrical phenomenon at the ESI capillary tip, can cause signal instability, increased chemical noise, and the degradation of labile analytes. [57] This guide provides an in-depth examination of these issues, exploring their fundamental mechanisms and presenting validated experimental strategies for their mitigation, framed within the broader context of robust ESI method development.
Salt Adduct Formation: Mechanisms and Mitigation Strategies
Underlying Mechanism and Contributing Factors
Salt adduct formation occurs when cations other than protons (e.g., Na+, K+) or anions associate with the analyte molecules during the ionization process. This results in heterogeneous ion populations visible in the mass spectrum as peak series corresponding to [M + zH + n(Na - H) + m(Cl + H)]z+, rather than a clean series of [M + zH]z+ ions. [55] This heterogeneity reduces spectral clarity, complicates data interpretation, and can diminish sensitivity by distributing the signal across multiple species.
The formation of these adducts is intrinsically linked to the mechanism of ESI. Two primary models explain the final production of gas-phase ions: the Charged Residue Model (CRM) for large molecules like folded proteins, and the Ion Evaporation Model (IEM) for smaller ions. [1] In both pathways, the presence of non-volatile salts in the solution means that these species are concentrated in the shrinking droplets alongside the analytes. If these salts are not adequately removed before the final ion release, they become incorporated into the gaseous analyte ion. The degree of adduction depends on several factors, including the concentration and type of salt, the physicochemical properties of the analyte, and the efficiency of droplet desolvation. [54] [55]
Experimental Characterization of Salt Effects
Research has dissected salt interference into two distinct aspects: adduct formation and ion suppression, which can occur independently. [55] A seminal study investigated the behavior of proteins under native and denaturing conditions in the presence of NaCl, CsCl, and tetrabutyl ammonium chloride (NBu4Cl). The key findings are summarized in Table 1.
Table 1: Experimental Characterization of Salt Effects on Protein ESI-MS
| Salt Type | Effect on Spectral Quality | Effect on Protein Ion Yield | Primary Mechanism |
|---|---|---|---|
| Sodium Chloride (NaCl) | Severe degradation via heterogeneous adduct formation ([M + zH + n(Na - H) + m(Cl + H)]z+). | Surprisingly robust integrated protein ion intensity. [55] | Adduct formation without significant suppression of ion yield; does not interfere with droplet fission. [55] |
| Tetrabutyl Ammonium Chloride (NBu4Cl) | No adduct formation. | Dramatic reduction in protein ion yield. [55] | Ion suppression; high surface affinity of NBu4+ reduces droplet charge and suppresses Rayleigh fission, lowering ion yield. [55] |
| Cesium Chloride (CsCl) | Combination of adduct formation and signal suppression. [55] | Reduced protein ion intensity. | Combination of both adduction and suppression effects. |
Detailed Experimental Protocol: [55]
- Sample Preparation: Protein solutions are prepared under both native (e.g., aqueous ammonium acetate) and denaturing (e.g., water/acetonitrile with formic acid) conditions.
- Salt Addition: Salts of interest (e.g., NaCl, CsCl, NBu4Cl) are added to the protein solutions at varying concentrations (e.g., 0-500 µM).
- ESI-MS Analysis: Samples are infused directly into the mass spectrometer using standard ESI conditions.
- Data Analysis: Mass spectra are analyzed for:
- Adduct Formation: Presence and pattern of additional peaks corresponding to sodium, cesium, or other adducts.
- Ion Suppression: Change in the total integrated signal intensity for all protein-related ions (e.g., [M+zH]z+ and adducted forms) compared to a salt-free control.
- Signal-to-Noise (S/N) Deterioration: Overall reduction in spectral quality.
Supporting Computational Methodology: [55] Molecular dynamics (MD) simulations of water droplets charged with Na+ or NBu4+ provide mechanistic insights. These simulations track droplet evolution, including charge-to-radius ratios (z/zR). Droplets with Na+ evolve close to the Rayleigh limit (z/zR ≈ 0.74), promoting fission, whereas NBu4+ with high surface affinity leads to lower z/zR values (~0.59), suppressing fission and reducing ion yield.
Diagram 1: Mechanism of Salt Adduct Formation in ESI.
Mitigation Strategies and Protocols
Several practical strategies can be employed to minimize salt adduction:
- Sample Desalting/Purification: This is the most effective method.
- Protocol: Use centrifugal filter devices with appropriate molecular weight cut-offs, solid-phase extraction (SPE) cartridges, or online liquid chromatography (LC) coupled to MS. For LC-ESI-MS, a trapping column can be used to capture and wash analytes before elution into the mass spectrometer.
- Mobile Phase Optimization:
- Protocol: Use volatile buffers such as ammonium acetate or ammonium formate (typically 1-50 mM) instead of non-volatile salts like phosphate buffers. Add volatile modifiers like 0.1% formic acid or acetic acid to enhance ionization and protonation. [1]
- ESI Source Parameter Adjustment:
- Protocol: Increase source temperature and drying gas flow rates (e.g., nitrogen) to enhance droplet desolvation, promoting the release of clean ions. Increase skimmer or cone voltage to induce collision-induced dissociation (CID) of weakly bound adducts in the source region (in-source CID). Caution: This may cause fragmentation of labile analytes.
- Employing Low-Flow ESI Techniques:
- Rationale: Techniques like nanoESI generate smaller initial droplets, which improves ionization efficiency and reduces nonspecific adducts. [58] FemtoESI, an ultra-low flow rate variant, has shown better desolvation and preservation of native protein structures. [58]
- Protocol: Utilize pulled glass or silica capillaries with tip diameters of 1-3 µm for nanoESI to achieve flow rates in the nanoliters per minute range. [1]
Ion Suppression in ESI: Matrix Effects and Solutions
Fundamental Causes and Gas-Phase Processes
Ion suppression is a matrix effect where the ionization efficiency of a target analyte is reduced due to the presence of other compounds (the matrix) in the sample. [54] [56] This leads to diminished signal intensity, poor quantitative accuracy, and can even result in false negatives. The mechanisms occur in both the liquid and gas phases.
In the liquid phase, suppression is primarily due to competition for available charge during droplet formation and fission. As droplets evaporate, analytes with higher surface activity or ionization efficiency may preferentially incorporate into the droplet surface and absorb a disproportionate share of the available charges. [54] The presence of non-volatile or less volatile compounds (e.g., salts, polymers, phospholipids) can also impede solvent evaporation or co-precipitate with the analyte, physically preventing its release. [56]
Crucially, studies have confirmed that gas-phase processes also dominate in certain types of ion suppression. [56] In Secondary Electrospray Ionization (SESI), for example, ion suppression has been strongly linked to gas-phase acid-base chemistry. An abundant molecule with high proton affinity (e.g., pyridine) can "steal" protons from a pre-formed, protonated analyte ion or from charged clusters in the gas phase, leading to signal reduction for the less basic analyte. [56] One proposed mechanism for SESI involves ligand switching in charged water clusters, where an abundant molecule displaces a lower-abundance molecule from the cluster. [56]
Experimental Analysis of Ion Suppression
A systematic study on SESI provides a robust protocol for characterizing ion suppression. [56]
Experimental Protocol for Assessing Ion Suppression: [56]
- Gas Standard Generation: Use an evaporation-based system to generate precise concentrations of gaseous analytes (e.g., acetone, deuterated acetone, deuterated acetic acid, pyridine) based on Henry's law. A dilution gas flow (e.g., 8 L/min) is used under controlled dry (0% RH) or humid (95% RH) conditions.
- Crossover Experiment Design:
- Maintain a constant concentration of one compound (e.g., D6-acetone at 0.011 ppm).
- Systematically increase the concentration of a competing compound (e.g., acetone) over a broad range (e.g., 1000x increase in half-logarithmic steps).
- Repeat, swapping the roles of the constant and variable compounds.
- SESI-MS Analysis: Analyze the gas stream using a SESI source coupled to a high-resolution mass spectrometer. Key settings: spray voltage +3.5 kV, electrolyte 0.1% formic acid, ionization chamber at 90°C.
- Data Acquisition: Operate the mass spectrometer in selected ion monitoring (SIM) mode to monitor the [M+H]+ ions of the target compounds (e.g., m/z 59 for acetone, m/z 64 for D6-acetone) to maximize sensitivity and minimize space charge effects.
- Data Analysis: Plot the signal intensity of the constant compound against the concentration of the variable compound. A significant decrease in signal indicates ion suppression.
Key Findings: [56]
- Pyridine exhibited the most significant suppressive effect, linked to its high gas-phase basicity.
- At an acetone concentration of 1 ppm (relevant to breath analysis), a ~30% decrease in intensity was observed for several features in a complex sample under humid conditions.
- Gas-phase effects were found to be a dominant factor in SESI ion suppression.
Diagram 2: Pathways of Ion Suppression in Liquid and Gas Phases.
Strategies to Overcome Ion Suppression
Mitigating ion suppression is critical for reliable quantification.
- Sample Clean-up:
- Protocol: Implement protein precipitation, liquid-liquid extraction, or solid-phase extraction (SPE) to remove interfering matrix components, particularly salts, phospholipids, and proteins, prior to LC-ESI-MS analysis. [54]
- Chromatographic Separation:
- Protocol: Develop LC methods to separate the target analyte from suppressing matrix components in the time domain. Use ultra-high-performance liquid chromatography (UHPLC) for superior peak capacity and resolution. Even a short separation can significantly reduce suppression compared to flow injection analysis. [54]
- Standardization:
- Protocol: Use stable isotope-labeled internal standards (SIL-IS) for each analyte. The SIL-IS co-elutes with the native analyte and experiences nearly identical suppression, allowing for accurate correction of the final quantified value. This is the gold standard for quantitative LC-ESI-MS.
- Methodological Dilution:
- Protocol: If sensitivity allows, dilute the sample to a point where the concentration of the suppressing matrix is below its effective threshold. This requires a prior investigation of the suppression profile.
Electrical Discharge and Its Interference
Origins and Consequences
Electrical discharge (or corona discharge) in ESI is an uncontrolled electrical phenomenon where the high electric field at the capillary tip ionizes the surrounding gas (typically air), creating a plasma. [57] This is generally considered detrimental to ESI performance. While it is intentionally harnessed in Atmospheric Pressure Chemical Ionization (APCI), in ESI it leads to several problems: [57]
- Signal Instability and Degradation: Degradation of the [M+H]+ ion intensity and increased chemical noise. [57]
- Electrochemical Complications: The discharge provides an alternative pathway for electron removal from the capillary tip, which can alter the electrochemical processes inherent to ESI and lead to the formation of unexpected species. [57]
- Oxidative Damage: The plasma can generate reactive oxygen species that may oxidize and damage labile analytes.
Discharge is more likely to occur under certain conditions, including the use of high capillary voltages, solvents with high surface tension (like pure aqueous buffers), and in the absence of a stable conductive Taylor cone, often at low flow rates. [57]
Experimental Observation and Controlled Utilization
A study deliberately maximized CD to develop a corona discharge-initiated electrochemical (CD-EC) ESI technique. [57]
Experimental Protocol for CD-EC-ESI: [57]
- Ion Source Configuration: A standard Z-Spray orthogonal atmospheric pressure ESI source was used with key modifications:
- The stainless steel capillary was extended 3 mm beyond the desolvation tube.
- Capillary voltage was maximized (5 kV).
- Desolvation temperature was maximized (500 °C).
- The angle and distance of the capillary relative to the cone were adjusted to maximize observable CD.
- A 33 MOhm current-limiting resistor was placed between the ESI capillary and the high-voltage supply.
- Analyte Selection: Ferrocene-labeled compounds (e.g., N-(Ferrocenyl)iodoacetamide, Fc-IAA) were used as electrochemically active model compounds with low ionization potential.
- Analysis: Samples were introduced via infusion or RP-HPLC. The mass spectrometer was monitored for the appearance of electrochemically generated radical cations ([M]•+) of the ferrocene compounds, which indicated successful CD-EC ionization.
- Current Measurement: A microammeter was used to measure current, with currents in excess of 10^-6 A indicative of CD conditions.
Findings: This setup created a robust electrochemical cell at the ESI tip, enabling efficient oxidation of analytes with high selectivity and sensitivity (up to zeptomolar levels). [57] This demonstrates that while typically unwanted, discharge can be harnessed for specific applications, though it fundamentally changes the ionization mechanism from standard ESI.
Mitigation of Unwanted Discharge
For conventional ESI-MS, preventing discharge is key.
- Solvent Modification:
- Protocol: Add modifiers to increase the solution's conductivity and reduce surface tension. A common mixture is water/organic solvent (e.g., methanol, acetonitrile) with 0.1-1.0% formic acid. [1] This facilitates stable Taylor cone formation at a lower voltage.
- Source Parameter Optimization:
- Protocol: Reduce the capillary voltage to the minimum required for stable spray. Use a heated drying gas (nitrogen) at optimal flow rates to aid desolvation without necessitating excessive voltages.
- Gas-Assisted Nebulization:
- Protocol: Many commercial ESI sources use a coaxial sheath gas (often nitrogen) to stabilize the spray and displace the ambient air, which can be prone to breakdown, from the high-field region.
- Use of Conductive Capillaries and Proper Grounding:
- Protocol: Ensure the ESI capillary is well-grounded and that all source components are properly aligned. Using emitters with thin, conductive coatings (e.g., gold) for nanoESI can help. [1]
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagents and Materials for Mitigating ESI Issues
| Item Name | Function/Benefit | Example Application |
|---|---|---|
| Ammonium Acetate/Formate | Volatile salt; replaces non-volatile buffers to minimize salt adduction. [54] | LC-ESI mobile phase for native MS or metabolomics. |
| Formic Acid/Acetic Acid | Volatile ion-pairing agent; enhances protonation, improves spray stability. [1] | Mobile phase modifier (0.1%) for positive ion mode ESI. |
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Corrects for ion suppression and variable recovery during sample preparation. [56] | Absolute quantification in bioanalytical LC-ESI-MS assays. |
| Solid-Phase Extraction (SPE) Cartridges | Sample clean-up; removes salts, phospholipids, and proteins causing suppression. [54] | Pre-treatment of plasma/serum samples before analysis. |
| Centrifugal Filter Devices | Rapid desalting and buffer exchange for protein/peptide samples. | Purifying a protein digest before nanoESI analysis. |
| NanoESI Capillaries | Low-flow ionization; reduces initial droplet size, improving sensitivity and reducing adducts. [58] [1] | High-sensitivity analysis of low-abundance protein digests. |
| Methanol/Acetonitrile (LC-MS Grade) | Organic modifiers; reduce surface tension, enhance desolvation, prevent discharge. [1] | Standard component of LC-ESI mobile phases. |
Integrated Workflow and Concluding Remarks
Successfully mitigating common ESI issues requires an integrated approach, from sample preparation to instrumental analysis. The following workflow diagram summarizes the key decision points and strategies.
Diagram 3: Integrated ESI-MS Workflow for Issue Mitigation.
In conclusion, the challenges of salt adducts, ion suppression, and electrical discharge are inherent yet manageable aspects of ESI-MS. A deep understanding of their underlying mechanisms—informed by studies employing molecular dynamics and systematic gas-phase experiments—is the foundation for developing robust analytical methods. By implementing a combination of strategic sample clean-up, chromatographic separation, careful solvent selection, and source parameter optimization, these issues can be effectively mitigated. As ESI-MS continues to be a pivotal tool in drug development and life sciences research, mastering these aspects of method development is paramount for generating high-quality, reliable data. The ongoing development of even softer and more efficient ionization methods, such as femtoESI, promises to further push the boundaries of what is analyzable. [58]
Solvent and Mobile Phase Selection to Enhance Ionization Efficiency and Signal Stability
In electrospray ionization (ESI), the formation of gas-phase ions from analyte molecules in solution is a critical process that underpins the sensitivity of Liquid Chromatography-Mass Spectrometry (LC-MS). The selection of solvents and the composition of the mobile phase are not merely chromatographic considerations; they are fundamental to the ionization mechanism itself. This guide details how strategic optimization of these parameters, within the broader context of understanding how electrospray ionization works, directly enhances ionization efficiency and ensures signal stability, leading to more robust and sensitive analytical methods for researchers and drug development professionals.
Core Principles of ESI and the Role of the Mobile Phase
The ESI Mechanism: From Droplet to Gas-Phase Ion
Electrospray ionization operates through a series of controlled physical processes that ultimately liberate analyte ions from a liquid matrix into the gas phase for mass spectrometric analysis [59]. The process begins when a high voltage is applied to a liquid passing through a capillary, dispersing it into a fine spray of charged droplets. As these droplets travel towards the mass spectrometer inlet, the solvent evaporates, increasing the charge density on the droplet surface. Repeated Coulombic fissions occur, breaking the droplets into progressively smaller offspring droplets. Ions are ultimately liberated into the gas phase via two postulated mechanisms: the Charge Residue Model (CRM) for larger molecules, where continued solvent evaporation leaves a charged analyte molecule, or the Ion Evaporation Model (IEM) for smaller ions, where direct field-assisted desorption from the droplet surface occurs [59] [60]. The efficiency of every step in this cascade is profoundly influenced by the properties of the mobile phase.
Foundational Properties of an Effective Mobile Phase
The mobile phase in ESI-MS serves a dual purpose: achieving chromatographic separation and facilitating efficient ionization. Several interconnected physical and chemical properties must be balanced to optimize both functions.
- Surface Tension: Solvents with lower surface tension, such as methanol or isopropanol, facilitate the formation of a stable Taylor cone and allow for the production of smaller initial droplets at a lower applied voltage. This promotes more efficient desolvation and ion release. The addition of even 1-2% of a low-surface-tension solvent like isopropanol to a highly aqueous mobile phase can significantly improve electrospray stability and instrument response [48].
- Dielectric Constant and Conductivity: The mobile phase must possess sufficient conductivity to allow for charge separation at the capillary tip. Highly non-polar solvents like hexane or toluene, common in Normal-Phase Liquid Chromatography (NPLC), cannot support a stable electrospray and require modification for ESI-MS coupling [61].
- Volatility: Volatile solvents and additives are preferred as they evaporate readily from charged droplets during the desolvation process. Non-volatile components can accumulate and lead to ion suppression and contamination of the ion source [62].
Optimizing Mobile Phase Composition
Solvent Selection and Ratios
The choice of organic modifier is a primary factor in controlling analyte retention and ionization efficiency.
Table 1: Common HPLC Solvents and Their Properties in ESI-MS
| Solvent | Surface Tension (mN/m) | Common Use | ESI Compatibility | Key Considerations |
|---|---|---|---|---|
| Water | High | Base solvent for RP-LC | Moderate | High content requires higher spray voltage; can destabilize spray [48]. |
| Acetonitrile | Medium | RP-LC modifier | Excellent | Low viscosity; sharp chromatographic peaks; common choice for ESI [62]. |
| Methanol | Low | RP-LC modifier | Excellent | Low surface tension promotes stable spray; can enhance response for some analytes [48]. |
| Isopropanol | Low | NPLC modifier / Additive | Good | Very low surface tension; useful as a minor additive (~1-2%) to stabilize aqueous sprays [48]. |
Fine-tuning the ratio of water to organic solvent is critical. A higher organic modifier concentration typically accelerates the elution of hydrophobic compounds and generally improves ionization efficiency by reducing droplet surface tension and enhancing desolvation. However, for very hydrophobic analytes, a high organic content can lead to premature elution without sufficient resolution [62]. The optimal ratio is best determined empirically for a given analyte set.
pH and Additive Selection
The pH of the mobile phase is one of the most powerful tools for optimizing ESI sensitivity, particularly for ionogenic analytes (acids and bases).
- Principle of pH Control: The mobile phase pH should be adjusted to promote the analyte's ionization. For basic analytes analyzed in positive ion mode, the mobile phase pH should be at least two units below the analyte pKa to ensure protonation. For acidic analytes analyzed in negative ion mode, the pH should be at least two units above the analyte pKa to ensure deprotonation [48]. This maximizes the concentration of pre-formed ions in solution, leading to a stronger signal.
- Common Additives:
- Acids/Bases: Formic acid and acetic acid (0.1%) are standard for positive ion mode. Ammonium hydroxide or ammonium acetate buffer is used for negative ion mode. Volatile ammonium acetate buffer (e.g., 5-20 mM) is widely used to stabilize pH for both positive and negative ionization [62] [59].
- Ion-Pairing Reagents: Reagents like alkylamines (e.g., triethylamine) can be used to form complexes with acidic analytes like oligonucleotides, improving their retention in reversed-phase chromatography and aiding in their transfer to the droplet surface during ESI [63].
- Additive Concentration: A critical balance must be struck. While additives are necessary, high concentrations (typically above 50 mM for salts) can cause significant ion suppression and should be avoided. All additives must be volatile to prevent source contamination [62] [63].
Table 2: Selection Guide for Mobile Phase Additives
| Additive | Typical Concentration | Ionization Mode | Function | Notes |
|---|---|---|---|---|
| Formic Acid | 0.05 - 0.1% | Positive | Promotes [M+H]+ formation; lowers pH | Volatile; common first choice [62]. |
| Ammonium Acetate | 2 - 20 mM | Positive & Negative | Buffers mobile phase pH | Volatile salt; high concentrations can suppress signal [62] [63]. |
| Ammonium Formate | 2 - 20 mM | Positive & Negative | Buffers mobile phase pH | Alternative to ammonium acetate for MS/MS applications. |
| Acetic Acid | 0.1 - 1% | Positive | Weaker acid than formic acid | Can provide different selectivity [62]. |
Practical Method Development and Troubleshooting
Systematic Optimization of ESI Source Parameters
The mobile phase composition is intrinsically linked to the optimal settings of the ESI source. A systematic approach to parameter tuning is essential.
Figure 1: A sequential workflow for tuning ESI source parameters in conjunction with the selected mobile phase. The process is iterative until optimal signal-to-noise and stability are achieved [48] [59].
Key parameters to optimize include:
- Capillary/Sprayer Voltage: This potential difference between the capillary tip and the MS inlet is responsible for forming the electrospray. The optimal voltage is dependent on the mobile phase composition; more aqueous phases often require a higher voltage. Using excessively high voltages can lead to electrical discharge and analyte fragmentation [48] [64].
- Nebulizing and Drying Gas: The nebulizer gas (often nitrogen) helps constrain the initial droplet size. Its flow rate should be optimized with the mobile phase flow rate. The drying gas, typically heated, aids in solvent evaporation from the charged droplets. The temperature must be balanced to ensure full desolvation without thermally degrading labile analytes [59].
Addressing Common Mobile Phase and Solvent Challenges
- Coping with Non-ESI Friendly Solvents: For techniques like Normal-Phase LC (NPLC) that use non-polar solvents (e.g., hexane), two primary solutions exist:
- Make-up Solvent Addition: A post-column T-union or coaxial setup is used to introduce a polar, ESI-friendly solvent (e.g., methanol/water with 0.1% acid) into the column eluent. This provides the necessary conductivity and polarity for a stable electrospray [61].
- Ambient Ionization Techniques: Methods like Continuous-Flow Extractive Desorption ESI (CF-EDESI) nebulize the NPLC eluent and mix it with charged droplets from an ESI emitter, extracting and ionizing analytes directly from the non-polar stream [61].
- Minimizing Signal Suppression and Adduct Formation:
- Signal Suppression: Caused by co-eluting matrix components that compete for charge or droplet surface area. Mitigation strategies include improved sample clean-up, better chromatographic separation, and reducing the source temperature for labile compounds [59].
- Adduct Formation: The presence of metal ions (e.g., Na+, K+) can lead to [M+Na]+ or [M+K]+ adducts, complicating spectra. Using high-purity solvents and additives, along with plastic instead of glass vials, can minimize this. For some analyses, additives like ammonium formate can promote the formation of a uniform [M+NH4]+ adduct [48].
- Preventing In-Source Fragmentation: Applying excessive energy in the interface region (e.g., high "cone" or "fragmentor" voltages) can cause analytes to fragment before mass analysis. This "in-source fragmentation" can generate ions that are mis-annotated as real lipids or metabolites. Lowering these voltages is key to preserving the intact molecular ion [64].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for ESI-MS Mobile Phase Preparation
| Item | Function | Example Use Case & Rationale |
|---|---|---|
| LC-MS Grade Water | Base solvent for mobile phase. | Minimizes chemical noise and background signals caused by impurities in lower-grade water. |
| LC-MS Grade Solvents | Organic modifiers (Acetonitrile, Methanol). | Reduces non-volatile residues and UV-absorbing impurities that suppress analyte signal. |
| Volatile Buffers | pH control (Ammonium Acetate, Formate). | Provides buffering capacity while being easily removed in the ESI process, preventing source contamination. |
| Volatile Acids/Bases | Ionization control (Formic, Acetic Acid, Ammonium Hydroxide). | Promotes analyte protonation/deprotonation without leaving non-volatile salts in the source. |
| Ion-Pairing Reagents | Modifies retention/ionization of charged analytes. | E.g., Alkylamines (TEA, HFIP) for oligonucleotide analysis; helps retain and ionize highly polar acids [63]. |
| Make-up Solvent System | Enables NPLC-ESI-MS coupling. | A pump and T-union to introduce an ESI-friendly solvent post-column, allowing ionization of non-polar eluents [61]. |
Experimental Protocol: A Practical Guide to Mobile Phase Optimization
This protocol outlines a systematic approach to optimizing mobile phase conditions for a small molecule drug candidate and its metabolites using a Q-TOF mass spectrometer.
1. Define Initial Conditions:
- Column: Select a suitable UHPLC column (e.g., C18, 100 x 2.1 mm, 1.7 µm).
- Mobile Phase A: Water with 0.1% formic acid.
- Mobile Phase B: Acetonitrile with 0.1% formic acid.
- Flow Rate: 0.4 mL/min.
- Injection: 5 µL of a 1 µg/mL solution of the analyte in a compatible solvent.
2. Scouting Gradient and Polarity:
- Run a generic, broad gradient (e.g., 5% B to 95% B over 10 minutes) to determine the approximate retention time of the analytes.
- Acquire data in both positive and negative ionization modes to determine the optimal polarity for each analyte.
3. Fine-Tuning Organic Modifier and pH:
- Modifier Scouting: Compare methanol versus acetonitrile as Mobile Phase B. Keep the acid concentration constant (0.1% formic acid). Observe changes in retention time, peak shape, and signal intensity.
- pH Scouting: For a basic analyte in positive mode, compare 0.1% formic acid (low pH) against 10 mM ammonium acetate (pH ~6.8) and 10 mM ammonium acetate with 0.1% ammonium hydroxide (higher pH). For acidic analytes in negative mode, compare ammonium acetate buffers with and without a basic modifier. The condition that yields the highest signal intensity for the protonated [M+H]+ or deprotonated [M-H]- ion is optimal.
4. Optimize Additive Concentration:
- If a buffer is chosen, perform a concentration study (e.g., 2 mM, 5 mM, 10 mM, 20 mM ammonium acetate). Inject the analyte and monitor the signal-to-noise ratio. Select the concentration that provides the best sensitivity without leading to long-term source contamination.
5. Link to Source Parameter Optimization:
- Once the mobile phase is defined, use the workflow in Figure 1 to fine-tune the ESI source parameters (voltage, gas flows, temperatures) for the specific analyte and mobile phase combination. This can be done manually or using automated Design of Experiments (DoE) approaches for multi-parameter optimization [47].
The path to achieving superior ionization efficiency and signal stability in ESI-MS is paved with informed and deliberate choices regarding the solvent and mobile phase. This process extends beyond mere chromatographic resolution to the very heart of the ionization mechanism. By understanding the core principles of ESI, methodically selecting and optimizing solvents, pH, and additives, and systematically linking these to source parameter tuning, scientists can develop robust, sensitive, and reliable LC-MS methods. This structured approach to mobile phase selection is fundamental to advancing research and accelerating drug development.
ESI in Context: Validation Frameworks and Comparison with Alternative Ionization Techniques
Cross-Calibration and Validation in Complex Biological Matrices like Whole Blood, Plasma, and Serum
In the realm of pharmaceutical development and bioanalysis, the generation of reliable pharmacokinetic (PK) data is paramount. This often necessitates the use of a validated bioanalytical method in more than one laboratory or the transition of a method to a new technological platform during a drug's development lifecycle. Cross-validation is a formal assessment process that demonstrates the equivalency of two or more validated bioanalytical methods [65]. Its fundamental purpose is to ensure that data generated from different sources are comparable and reliable, a critical requirement when pooling data from multiple clinical sites for global studies.
This process is intricately linked to the broader research on electrospray ionization (ESI), which is a cornerstone of modern liquid chromatography-tandem mass spectrometry (LC-MS/MS). ESI is a complex mechanism involving the nebulization of a sample solution into charged droplets, followed by the liberation of ions from these droplets into the gas phase for mass analysis [66]. The composition of the biological matrix—such as whole blood, plasma, or serum—can significantly influence the efficiency and reproducibility of the ESI process. Understanding these mechanistic aspects is therefore essential for developing robust analytical methods, as the matrix can affect ionization efficiency, a phenomenon influenced by analyte dimension, charge state, and ion mobility in solution [67]. Consequently, cross-validation ensures that despite the complexities introduced by biological matrices and the ESI process, the final analytical results remain consistent and accurate across different laboratories and methodologies.
Theoretical Foundations and Regulatory Context
Principles of Cross-Validation
Cross-validation is defined as an experimental procedure to demonstrate the equivalence of two or more validated bioanalytical methods [65]. Its strategic importance lies in its ability to safeguard data integrity when methods are transferred between laboratories or when a method undergoes a significant platform change. This is a common occurrence as drug development programs progress from early to late stages, often expanding globally and requiring more advanced analytical techniques.
The primary objective is to ensure that the concentration of an analyte (e.g., a drug) measured in a biological sample is equivalent, regardless of which validated method or laboratory is used. This is crucial because pharmacokinetic parameters derived from these concentrations directly inform decisions about drug safety and efficacy. The current health agency guidance provides detailed recommendations for the initial validation of bioanalytical methods within a single laboratory, but guidance on cross-validation is more limited, leading organizations to develop their own robust, science-driven strategies [65].
The Electrospray Ionization (ESI) Link
The cross-validation of LC-MS/MS methods is deeply connected to the fundamentals of electrospray ionization. ESI is a complex process that can be divided into three main steps:
- Nebulization: A sample solution is fed through a capillary, and a high electric field pulls charge towards the liquid front, forming a Taylor cone and emitting charged droplets [66].
- Droplet Disintegration and Evaporation: The charged droplets travel through the surrounding gas, undergoing solvent evaporation and Coulombic fissions, leading to progressively smaller droplets with higher charge density [66].
- Ion Liberation: When the electric field at the droplet surface becomes sufficiently high, gas-phase ions are emitted from the droplets and sampled by the mass analyzer [66].
The "matrix effect" in ESI is a well-known challenge. The chemical composition of the sample solution, including the biological matrix and its constituents, can strongly affect the ionization efficiency of the analyte [67]. Components from plasma or serum can suppress or enhance ionization, leading to inaccurate quantification. The sample ion intensity is dependent on ion structure and is affected by solvent composition and the presence of additives [66]. Cross-validation studies are, therefore, critical to confirm that different sample preparation techniques (e.g., protein precipitation, liquid-liquid extraction, solid-phase extraction) used across laboratories effectively and consistently mitigate these matrix effects, ensuring that the ESI process yields comparable results.
Experimental Design and Methodologies
A well-designed cross-validation study is built on a foundation of rigorous planning and statistical foresight. The strategy developed at Genentech, Inc., which utilizes incurred samples (real study samples from dosed subjects), provides a robust framework for these assessments [65].
Core Experimental Strategy
The protocol involves selecting approximately 100 incurred study samples that span the applicable range of concentrations. These samples are strategically chosen based on four quartiles (Q) of in-study concentration levels to ensure the entire analytical range is evaluated. Each of these samples is assayed once by the two bioanalytical methods being compared [65].
The statistical analysis for establishing equivalency is pre-specified. The two methods are considered equivalent if the percent differences in the lower and upper bound limits of the 90% confidence interval (CI) for the 100 samples are both within ±30% [65]. To provide a more granular view, a quartile-by-concentration analysis using the same acceptability criterion may also be performed. Furthermore, a Bland-Altman plot—which graphs the percent difference of sample concentrations against the mean concentration of each sample—is created to help visualize the agreement between the two methods and identify any concentration-dependent biases [65].
Case Study: Inter-Laboratory Cross-Validation for Lenvatinib
A comprehensive inter-laboratory cross-validation study for lenvatinib, a novel multi-targeted tyrosine kinase inhibitor, exemplifies this process in practice [68]. Five laboratories across Asia, the US, and Europe developed seven distinct LC-MS/MS methods for quantifying lenvatinib in human plasma. Each method was first individually validated according to bioanalytical guidelines before the cross-validation was initiated.
Table 1: Summary of Lenvatinib Bioanalytical Methods Used in Cross-Validation [68]
| Laboratory | Method | Assay Range (ng/mL) | Sample Extraction | Internal Standard (IS) | Chromatography Column |
|---|---|---|---|---|---|
| A (Asia) | A | 0.1 - 500 | LLE by diethyl ether | ER-227326 | Symmetry Shield RP8 |
| B (US) | B | 0.25 - 250 | PP by ACN-MeOH | 13C6 lenvatinib | Hypersil Gold |
| C (US) | C | 0.25 - 250 | LLE by MTBE-IPA | 13C6 lenvatinib | Synergi Polar-RP |
| D (EU) | D | 0.1 - 100 | LLE by diethyl ether | ER-227326 | Symmetry Shield RP8 |
| E (Asia) | E1, E2, E3 | 0.25 - 500 / 0.25 - 250 | SPE / LLE / SPE | ER-227326 / 13C6 lenvatinib | Synergi Polar-RP / Luna C18(2) |
In this study, quality control (QC) samples and blinded clinical study samples were exchanged and analyzed. The results demonstrated a high level of comparability, with the accuracy of QC samples within ±15.3% and the percentage bias for clinical study samples within ±11.6% [68]. These findings confirmed that lenvatinib concentrations in human plasma could be reliably compared across different laboratories and clinical studies, despite variations in specific methodological details.
The following workflow diagram illustrates the key stages of a typical cross-validation process for bioanalytical methods:
The Scientist's Toolkit: Essential Reagents and Materials
The successful execution of a bioanalytical method, particularly one relying on ESI-MS, depends on a suite of high-quality reagents and materials. The following table details key components used in the featured lenvatinib study and their critical functions [68].
Table 2: Key Research Reagent Solutions for LC-MS/MS Bioanalysis [68]
| Item | Function in the Analytical Process |
|---|---|
| Analyte Reference Standard | Provides the authentic compound for creating calibration curves and quality control samples, ensuring accurate quantification. |
| Internal Standard (IS) | Corrects for variability in sample preparation and ionization; can be a structural analogue (e.g., ER-227326) or a stable isotope-labeled version (e.g., 13C6-lenvatinib). |
| Blank Biological Matrix | Drug-free human plasma, serum, or whole blood from appropriate sources, used to prepare calibrants and QCs that mimic the study samples. |
| Sample Extraction Solvents | Chemicals for protein precipitation (ACN, MeOH), liquid-liquid extraction (Diethyl ether, MTBE), or solid-phase extraction (SPE plates) to isolate the analyte from the matrix. |
| Mobile Phase Additives | Volatile acids (Formic Acid, FA) and buffers (Ammonium Acetate, NH4Ac) that facilitate chromatography and enhance ESI ionization efficiency. |
| Chromatography Column | The stationary phase (e.g., C8, C18, Polar-RP) where the physical separation of the analyte from matrix components occurs. |
Detailed Experimental Protocols
Sample Preparation Techniques
The lenvatinib cross-validation study showcased three primary sample preparation techniques, each with its own protocol [68]:
- Protein Precipitation (PP): For method B, a 0.05 mL plasma sample was mixed with a 0.3 mL solution of acetonitrile and methanol (2:1 ratio) containing the internal standard. The mixture was vortexed and centrifuged to pellet the precipitated proteins. The supernatant was then collected for analysis [68].
- Liquid-Liquid Extraction (LLE): In methods A, C, and D, a plasma aliquot (0.1-0.2 mL) was mixed with an organic solvent. For method A, 2.5 mL of diethyl ether was used. For method C, 0.75 mL of methyl tert-butyl ether and isopropanol (7:3) with 0.1% acetic acid was employed. The mixture was shaken and centrifuged to separate the phases. The organic (top) layer, containing the extracted analyte, was transferred and evaporated to dryness. The residue was then reconstituted in an injection-compatible solvent [68].
- Solid Phase Extraction (SPE): Method E1 used a hydrophilic-lipophilic balanced (HLB) SPE plate. The plasma sample was loaded onto the preconditioned plate, washed to remove impurities, and then the analyte was eluted with a strong solvent, such as 0.4 mL of 0.2 mM ammonium acetate in acetonitrile with 1% formic acid [68].
LC-MS/MS Analysis Protocol
The general LC-MS/MS analysis follows a standardized workflow, with specific details from the lenvatinib methods provided as examples [68]:
- Chromatography: The reconstituted sample extract is injected onto a reversed-phase HPLC column. The lenvatinib methods used columns like Symmetry Shield RP8 and Synergi Polar-RP. The analyte is separated using a mobile phase gradient, typically composed of an aqueous component (A) such as 0.1% formic acid and an organic component (B) such as methanol or acetonitrile [68].
- Ionization: The eluent from the HPLC column is introduced into the ESI source, which is operated in positive ion mode for lenvatinib. Here, the analyte is nebulized into charged droplets and converted into gas-phase ions [66].
- Mass Analysis: The gas-phase ions enter the mass spectrometer. The instrument is set to Multiple Reaction Monitoring (MRM) mode, where it specifically selects the precursor ion of lenvatinib, fragments it in a collision cell, and then monitors for a specific product ion. This two-stage mass selection provides high specificity and sensitivity [68].
- Data Processing: The peak areas for the analyte and internal standard are recorded. The analyte-to-IS peak area ratio is calculated and compared against the calibration curve to determine the absolute concentration of the analyte in the original sample.
The relationship between the sample matrix, sample preparation, and the ESI process is complex and critical to the success of the method, as summarized in the following diagram:
Data Analysis and Acceptance Criteria
The final step in cross-validation is a rigorous statistical evaluation of the generated data to determine method equivalency.
- Primary Acceptance Criterion: As per the Genentech strategy, the two methods are considered equivalent if the 90% confidence interval (CI) limits for the mean percent difference of concentrations from about 100 incurred samples fall entirely within ±30% [65].
- Supplementary Analysis: A quartile analysis is often performed to ensure that this acceptability criterion is met across different concentration levels (low, medium, high). This helps identify any potential bias at the extremes of the calibration range.
- Data Visualization: The Bland-Altman plot is an invaluable tool for visually assessing the agreement between the two methods. It plots the percent difference between the two methods against the average concentration of each sample. This plot can reveal systematic biases (e.g., one method consistently reading higher than the other) or trends where the disagreement between methods changes with concentration [65].
In the lenvatinib study, the success of the cross-validation was demonstrated by the fact that all accuracy and bias results for QC and study samples were well within the pre-defined acceptance criteria, proving that the various methods, despite their differences in sample preparation and chromatography, produced comparable results [68].
Within the broader context of research into how electrospray ionization works, understanding the relative strengths and weaknesses of different ionization techniques is fundamental to advancing mass spectrometry. Electrospray Ionization (ESI) and Atmospheric Pressure Photoionization (APPI) represent two powerful but distinct soft ionization methods that have revolutionized the analysis of non-volatile and thermally labile compounds. While ESI has become the default technique for a wide array of applications, particularly in the life sciences, APPI has emerged as a vital complementary technique for compounds that are poorly ionized by ESI. This in-depth technical guide examines the fundamental ionization mechanisms, comparative performance characteristics, and application suitability of ESI and APPI, providing researchers and drug development professionals with a structured framework for selecting the optimal ionization source for their analytical challenges. The distinct ionization pathways of these techniques—one driven by electric fields and charged droplet formation, the other by photon energy and gas-phase reactions—underpin their unique performance characteristics and application domains [69] [70].
Fundamental Ionization Mechanisms
Electrospray Ionization (ESI) Mechanism
Electrospray Ionization operates at atmospheric pressure and begins when a sample solution containing the analyte is passed through a charged metal capillary (typically applied with 3-5 kV). This high voltage induces a charge on the liquid, creating a Taylor cone from which a fine spray of charged droplets emerges. As these charged droplets travel towards the mass spectrometer inlet, the solvent evaporates with the assistance of a nebulizing gas and heat, causing the droplets to shrink while increasing their charge density. When the Rayleigh limit is reached—the point where Coulombic repulsion overcomes the droplet's surface tension—the droplets undergo Coulombic fission, dividing into smaller droplets. This process repeats until completely desolvated analyte ions are released into the gas phase, ready for mass analysis [71] [70] [72].
A key characteristic of ESI is its ability to produce multiply charged ions, particularly for large biomolecules such as proteins and peptides. This multiple charging phenomenon effectively reduces the mass-to-charge ratio (m/z) of high molecular weight compounds, making them amenable to analysis by mass spectrometers with limited m/z ranges. The ionization process in ESI primarily occurs through the formation of protonated molecules [M+H]⁺ or deprotonated molecules [M-H]⁻ in positive and negative mode, respectively, though adduct formation with alkali metal ions (e.g., [M+Na]⁺) is also common. The efficiency of ESI is highly dependent on the analyte's ability to be charged in solution, making it particularly suitable for polar or ionic compounds that can be easily pre-charged in the liquid phase [71] [70].
Atmospheric Pressure Photoionization (APPI) Mechanism
Atmospheric Pressure Photoionization employs a fundamentally different approach based on photon energy rather than electrostatic forces. In APPI, the sample solution is first vaporized by a heated nebulizer to create a gas-phase aerosol. This aerosol is then exposed to photons from a vacuum ultraviolet (VUV) lamp, typically krypton (10.0 eV) or xenon (8.4 eV), which emits photons with sufficient energy to ionize suitable molecules. The ionization process can proceed through two primary pathways: direct photoionization and dopant-assisted photoionization [69] [73] [70].
In direct photoionization, analyte molecules (M) with ionization energies lower than the photon energy directly absorb photons and eject electrons to form molecular radical cations (M⁺•):
M + hν → M⁺• + e⁻
This direct mechanism is particularly effective for non-polar compounds with aromatic rings or conjugated systems that can stabilize the resulting radical cation. However, for many analytes, especially in complex solvent systems, a dopant-assisted mechanism (also called photo-induced chemical ionization) predominates. In this approach, a dopant compound (D) with high photoabsorption cross-section (e.g., toluene, acetone, or anisole) is added to the mobile phase. The dopant is first ionized by the VUV light:
D + hν → D⁺• + e⁻
The resulting dopant radical cations then react with analyte molecules through charge exchange or with solvent molecules to produce protonating agents that can protonate the analyte:
D⁺• + M → M⁺• + D (charge exchange)
D⁺• + S → [S+H]⁺ + D-H• (protonation via solvent)
The dopant-assisted mechanism significantly extends the range of compounds that can be effectively ionized by APPI, including many moderately polar molecules that might not ionize efficiently through direct photoionization [69] [73].
Comparative Ionization Pathways
The diagram below illustrates the fundamental ionization pathways for both ESI and APPI, highlighting their distinct mechanisms and common ionization products.
Critical Performance Comparison
Quantitative Analytical Figures of Merit
The distinct ionization mechanisms of ESI and APPI translate directly into different analytical performance characteristics. A comprehensive comparative study analyzing lipids demonstrated that APPI generally offered lower detection limits, higher signal intensities, and superior signal-to-noise ratios compared to ESI and APCI. Specifically, APPI was found to be 2-4 times more sensitive than APCI and significantly more sensitive than ESI without mobile-phase modifiers for neutral lipid classes [74]. Both APPI and APCI showed comparable linear ranges of 4-5 decades, while ESI sensitivity, though dramatically enhanced by mobile-phase modifiers such as ammonium formate or sodium acetate, often resulted in less stable adduct signals that were either nonlinear or had reduced linear ranges [74].
In lipidomic analysis, APPI has proven particularly valuable for non-polar and low-polarity lipids, offering the highest signal-to-noise ratio, sensitivity, repeatability, and the lowest limits of detection and quantification. Conversely, ESI and APCI remain better suited for analyzing polar lipids under normal-phase HPLC conditions. This polarity-dependent performance was clearly demonstrated in the analysis of Leishmania donovani parasites, where APPI excelled for non-polar lipids like squalene and cholesteryl esters, while ESI provided superior performance for polar phospholipids including lyso-phosphatidylcholine and phosphatidylglycerol [75].
Table 1: Comparative Analytical Performance of ESI and APPI
| Performance Parameter | Electrospray Ionization (ESI) | Atmospheric Pressure Photoionization (APPI) |
|---|---|---|
| Ionization Mechanism | Charge residue from charged droplets | Direct photoionization or dopant-mediated proton transfer |
| Ion Types Produced | [M+H]⁺, [M+Na]⁺, [M-H]⁻, multiply charged ions | M⁺•, [M+H]⁺, predominantly singly charged |
| Optimal Analyte Polarity | Medium to high polarity [76] | Low to medium polarity, non-polar [76] |
| Typical Flow Rates | Best with low flow rates (≤ 1000 μL/min) [77] | Works across a range, including high flow rates [73] |
| Matrix Effects | Susceptible to ion suppression from co-eluting compounds [69] | Generally more tolerant to matrix components [69] |
| Linear Dynamic Range | Reduced range with mobile-phase modifiers [74] | Wide linear range (4-5 decades) [74] |
| Relative Sensitivity | Lower for non-polar compounds without modifiers [74] | Higher for non-polar and moderately polar compounds [74] |
Analyte-Dependent Performance and Application Suitability
The complementarity between ESI and APPI becomes particularly evident when examining their performance across different compound classes. For environmentally relevant pharmaceuticals including antibiotics, beta-blockers, and selective serotonin reuptake inhibitors, most compounds ionized preferentially by ESI, though some performed significantly better using APPI [69]. This pattern highlights the value of having multiple ionization techniques available when developing methods for diverse compound panels.
In lipid analysis, ESI has demonstrated particular effectiveness in ionizing a broader range of cholesteryl esters compared to APCI. The ESI process generates strong signal intensity of precursor ions corresponding to [M+Na]⁺ and [M+NH₄]⁺ regardless of the number of carbon chains and double bonds in cholesteryl esters, while APCI selectively sensitizes detection of cholesteryl esters with unsaturated fatty acids, producing protonated ions [M+H]⁺ with weaker signal intensity [78]. This differential performance based on lipid class and fatty acid composition underscores the importance of matching the ionization technique to the specific analytical targets.
Table 2: Application-Based Selection Guide for ESI vs. APPI
| Analytical Application | Recommended Ionization Technique | Key Considerations |
|---|---|---|
| Pharmaceutical Analysis | ESI for polar APIs; APPI for non-polar compounds | ESI preferred for most pharmaceuticals; APPI valuable for specific non-polar drugs [69] |
| Lipidomics | ESI for polar lipids (phospholipids); APPI for neutral lipids (triglycerides, cholesteryl esters) | APPI provides superior sensitivity for non-polar lipid classes; ESI better for polar phospholipids [74] [75] |
| Environmental Analysis | Both, with compound-specific optimization | ESI suitable for most contaminants; APPI complementary for non-polar pesticides and emerging contaminants [69] |
| Metabolomics | Primarily ESI, with APPI for non-polar metabolites | Broad metabolite coverage often requires multiple ionization techniques |
| Petrochemical Analysis | Primarily APPI | APPI particularly effective for polyaromatic hydrocarbons (PAHs) and non-polar hydrocarbons [70] |
| Protein/Peptide Analysis | Exclusively ESI | APPI unsuitable for large, thermolabile biomolecules [69] [70] |
| Polymer Analysis | ESI for polar polymers; APPI for non-polar polymers | Charge state distribution in ESI enables molecular weight determination |
Experimental Protocols and Methodologies
Standard Operating Procedure for Comparative Ionization Studies
A robust experimental protocol for comparing ESI and APPI performance should begin with instrument configuration. Most modern mass spectrometers allow for relatively straightforward interchange between ESI and APPI sources, though some systems offer dual-mode sources that can operate in both modalities [73]. For ESI, typical operating parameters include: capillary voltage of 3-4 kV, vaporizer temperature of 200-300°C, sheath gas pressure of 20-50 arbitrary units, and auxiliary gas flow of 5-20 units. For APPI, the vaporizer temperature typically ranges from 300-500°C, with the VUV lamp (either krypton or argon) positioned optimally for maximum photon flux. Dopants such as toluene or acetone are commonly introduced at 0.1-0.5% v/v either via a separate infusion pump or premixed with the mobile phase [69] [73].
Chromatographic conditions must be optimized for compatibility with both ionization techniques. Normal-phase solvent systems are often employed for lipid analyses due to low solubility of these compounds in aqueous reversed-phase solvent systems [74]. For the analysis of lipids from Leishmania donovani parasites, a normal-phase separation was implemented using a monolithic silica column with common normal-phase solvents. The mobile phase typically consisted of hexane and methyl tert-butyl ether with ammonium acetate as an additive, which proved compatible with both ESI and APPI interfaces [75].
Method Optimization and Validation
Method optimization should follow a systematic approach, beginning with direct infusion of individual standards to determine optimal ionization conditions for each technique. For ESI, parameters such as capillary temperature, spray voltage, and gas flows should be optimized to maximize signal intensity while minimizing in-source fragmentation. Mobile phase additives (e.g., ammonium formate, sodium acetate, or acetic acid) can significantly enhance ESI sensitivity for certain compound classes but may reduce linear dynamic range [74].
For APPI optimization, both lamp type (krypton vs. argon) and dopant selection require careful evaluation. Krypton lamps (10.0 eV) generally provide better performance for direct photoionization, while argon lamps (8.4 eV) may enhance photo-induced chemical ionization for certain applications [73]. Dopants such as toluene, acetone, or anisole should be screened for their efficiency in promoting analyte ionization. The diagram below illustrates a systematic workflow for method development and optimization when comparing ESI and APPI techniques.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for ESI and APPI Experiments
| Category | Specific Items | Function and Application Notes |
|---|---|---|
| Mobile Phase Additives | Ammonium formate, ammonium acetate, sodium acetate, formic acid, acetic acid | Enhance ionization efficiency in ESI; concentration typically 1-10 mM [74] [78] |
| APPI Dopants | Toluene, acetone, anisole | Promote efficient charge transfer in APPI; typically used at 0.1-0.5% v/v [75] [73] |
| HPLC Solvents | Acetonitrile, methanol, water, propan-2-ol, chloroform, hexane | Solvent compatibility differs: ESI requires polar solvents; APPI tolerates normal-phase solvents [74] [76] |
| Reference Standards | Squalene, cholesteryl esters, triglycerides, phospholipids, pharmaceutical compounds | System suitability testing and method validation [75] [78] |
| Ion Source Components | ESI capillaries, APPI VUV lamps (krypton, argon), nebulizers, heating elements | Source maintenance and performance optimization |
ESI and APPI represent complementary ionization techniques with distinct mechanisms and application domains grounded in their fundamental physical principles. ESI excels for polar to moderately polar compounds, particularly large biomolecules that benefit from multiple charging, while APPI extends the analytical capability of LC-MS to non-polar and moderately polar compounds that are poorly ionized by ESI. The choice between these techniques should be guided by the physicochemical properties of the target analytes, with ESI remaining the default for most pharmaceutical and biomolecular applications, and APPI providing critical capabilities for lipidomics, environmental analysis, and petrochemical applications. As mass spectrometry continues to evolve, the strategic implementation of both techniques—either sequentially or through multimode sources—will empower researchers to address increasingly complex analytical challenges in drug development and biological research.
In the realm of mass spectrometry, the mass analyzer serves as the core component responsible for separating ions based on their mass-to-charge ratio (m/z). The selection of an appropriate mass analyzer is paramount for the success of experiments leveraging electrospray ionization (ESI), a soft ionization technique that efficiently produces ions from liquid samples, making it ideal for the analysis of biomolecules and pharmaceuticals. This technical guide provides an in-depth evaluation of four pivotal mass analyzer technologies: quadrupole, ion trap, Fourier Transform Ion Cyclotron Resonance (FT-ICR), and Quadrupole Time-of-Flight (Q-TOF). Each analyzer offers distinct capabilities in terms of mass resolution, accuracy, dynamic range, and tandem MS functionalities, making them uniquely suited for specific applications within drug development and related research fields. Framed within the context of ESI-based research, this whitepaper equips scientists with the knowledge to select the optimal analytical tool for their specific experimental needs, from routine quantification to the detailed structural elucidation of complex molecules.
Core Principles and Comparative Analysis of Mass Analyzers
Fundamental Operating Principles
Quadrupole Mass Analyzer: A quadrupole mass analyzer functions as a mass filter. It consists of four parallel, precisely machined metal rods. Opposing rod pairs are connected electrically, with one pair having a combined radio frequency (RF) and direct current (DC) voltage applied, and the other pair having an RF voltage 180 degrees out of phase with a superimposed DC voltage of opposite polarity [79] [80]. This configuration creates a dynamically oscillating electric field in the space between the rods. For a given set of RF and DC voltages, only ions of a specific m/z value will develop a stable trajectory and successfully traverse the full length of the quadrupole to reach the detector. All other ions, with unstable trajectories, will collide with the rods or be ejected radially from the analyzer [79] [81]. The mass spectrum is acquired by systematically varying (scanning) the applied voltages while maintaining a fixed ratio, thereby bringing different m/z values into a stable transmission window.
Quadrupole Ion Trap (QIT) Analyzer: The quadrupole ion trap operates on similar physical principles as the linear quadrupole but is configured to trap and store ions in a three-dimensional space. A typical QIT consists of a central ring electrode and two hyperbolic end-cap electrodes [82]. By applying appropriate RF potentials to these electrodes, a potential well is created, confining ions within the cavity of the trap. A damping gas, typically helium, is present at a relatively high pressure (~10⁻³ Torr) to cool the ions via collisions, concentrating them near the center of the trap and improving trapping efficiency [82]. Mass analysis is performed using the mass-selective instability scan: the amplitude of the RF voltage on the ring electrode is linearly increased, which sequentially destabilizes the trajectories of ions of increasing m/z. These unstable ions are ejected axially through openings in the end-cap electrodes toward the detector [82]. The QIT is a tandem-in-time instrument, as multiple stages of MS (MSⁿ) can be performed by isolating a specific precursor ion, fragmenting it via resonant excitation, and then analyzing the resulting product ions, all within the same physical space but in sequential time periods [82].
Fourier Transform Ion Cyclotron Resonance (FT-ICR) Mass Analyzer: FT-ICR MS is based on the principle of ion cyclotron resonance. Ions trapped in a powerful, static, and homogeneous magnetic field will travel in circular paths (cyclotron motion) perpendicular to the magnetic field lines. The frequency of this motion, the cyclotron frequency (ωc), is inversely proportional to the m/z value (ωc = k * B / m/z, where B is the magnetic field strength and k is a constant) [82] [83]. Ions of the same m/z move coherently but are not initially in phase. A broadband RF excitation pulse is applied at their cyclotron frequency, which synchronizes their motion into a coherent packet and increases their orbital radius. This coherently moving ion packet then induces an image current (a detectable time-domain signal) on a pair of detector plates as it passes close to them. This signal, called a transient, contains the frequency components of all the ions in the trap. The mass spectrum is obtained by applying a Fourier Transform to this time-domain signal, deconvoluting it into the individual frequency components, which are then converted to m/z values [83]. The ultra-high resolution and mass accuracy of FT-ICR stem from the precision with which these frequencies can be measured, directly benefiting from stronger magnetic fields.
Quadrupole Time-of-Flight (Q-TOF) Mass Analyzer: A Q-TOF is a hybrid instrument that combines a front-end mass-filtering quadrupole (Q) with a high-resolution time-of-flight (TOF) mass analyzer [84]. In a TOF analyzer, ions are accelerated by an electric field to a consistent kinetic energy, causing them to travel down a field-free drift tube. Since ions with the same energy but lower m/z travel faster than those with higher m/z, they reach the detector sooner. The m/z of an ion is determined by its time of flight [85] [81]. In the Q-TOF configuration, the quadrupole can be operated either in RF-only mode to transmit a broad m/z range or as a mass filter to select a specific precursor ion for MS/MS experiments. The selected ions are then fragmented in a collision cell (typically another RF-only quadrupole or hexapole), and all resulting product ions are pulsed into the TOF analyzer for simultaneous, high-resolution mass analysis [84]. This provides accurate mass data for both precursor and product ions, which is invaluable for structural elucidation and confident identification of unknowns.
Comparative Performance Data
The following table summarizes the key performance characteristics of the four mass analyzers, providing a quantitative basis for comparison. Data is compiled for systems using electrospray ionization, a common source for the analysis of pharmaceuticals and biomolecules.
Table 1: Comparative Performance Metrics of Mass Analyzers
| Analyzer Type | Mass Resolution (FWHM) | Mass Accuracy (ppm) | Dynamic Range | Tandem MS Capability | Optimal Application Context in ESI Research |
|---|---|---|---|---|---|
| Quadrupole | Unit resolution (1,000 - 4,000) [80] | ~100 ppm [80] | 4-5 orders of magnitude [85] | MS/MS (Tandem-in-space, e.g., Triple Quad) [79] [81] | Targeted quantification (SRM/MRM), GC/LC-MS coupling [79] [80] |
| Ion Trap (QIT) | Unit resolution (up to ~4,000) [82] | ~100 ppm | Limited by space charge [82] | MSⁿ (Tandem-in-time, multiple stages) [82] | Fragmentation pathway elucidation, structural studies of ions |
| Q-TOF | 15,000 - 60,000 [85] [81] | < 3 - 5 ppm (with lock mass) [85] | 4 orders of magnitude [85] | MS/MS (High-res product ion scanning) [84] | Untargeted screening, metabolite ID, peptide sequencing |
| FT-ICR | > 1,000,000 @ m/z 400 [83] | < 1 ppm (sub-ppm routine) [83] | > 500:1 per pixel (in MSI) [83] | MS/MS with ultra-high resolution | Unraveling complex mixtures, petroleomics, top-down proteomics |
Experimental Protocols and Workflows
Protocol for Targeted Quantification using a Triple Quadrupole Mass Spectrometer
This protocol details the use of a triple quadrupole (QqQ) mass spectrometer for Selected Reaction Monitoring (SRM), the gold standard for sensitive and specific quantification of target analytes, such as a drug and its metabolites in plasma.
1. Instrument Calibration and Tuning:
- Perform mass calibration using a standard tuning solution specific to the instrument manufacturer across the intended mass range.
- Optimize the ESI source parameters (nebulizer gas, drying gas, capillary voltage, source temperature) by infusing a standard solution of the analyte to maximize the intensity of the precursor ion.
- For the Q-Tof, perform a mass calibration in both V-mode (for robustness) and W-mode (for higher resolution) using a reference compound like leucine enkephalin, ensuring mass accuracy residuals are below 0.05 mDa [85].
2. Compound Optimization:
- Directly infuse a pure standard of the target analyte (e.g., Temazepam at 500 pg/µL) into the ESI source [85].
- Q1 MS Scan: With Q3 in RF-only mode, perform a Q1 scan to identify the intact precursor ion (e.g., [M+H]⁺).
- Product Ion Scan: Set Q1 to transmit only the selected precursor ion. Introduce collision gas (N₂ or Ar) into q2 and perform a product ion scan by ramping the collision energy (CE) in Q3 to generate a full fragmentation spectrum.
- SRM Optimization: For the most abundant and specific product ions, systematically optimize the collision energy for each transition to maximize the signal. Define the final SRM transition as Precursor m/z → Product m/z.
3. Liquid Chromatography Method Development:
- Develop a reversed-phase LC method to chromatographically separate the analyte from matrix interferences.
- Use a UPLC system with a C18 column (e.g., 2.1 x 100 mm, 1.7 µm) and a gradient of water and acetonitrile, both with 0.1% formic acid [85].
- Adjust the gradient profile and flow rate to achieve a sharp, symmetric peak for the analyte with a retention time that provides sufficient separation from the solvent front and other matrix components.
4. Data Acquisition and Analysis:
- Program the data acquisition method with the optimized SRM transitions and their corresponding retention time windows.
- For internal standardization, use a stable isotope-labeled version of the analyte.
- Acquire data for calibration standards (e.g., 0.5 pg/µL to 5000 pg/µL) and quality control samples in triplicate [85].
- Process data by integrating the peak areas for the SRM transitions. Generate a calibration curve (peak area vs. concentration) and determine the correlation coefficient (r), which should be ≥0.99, and the lower limit of quantification (LLOQ) [85].
Protocol for Untargeted Metabolomics using a Q-TOF Mass Spectrometer
This protocol outlines a non-targeted workflow for profiling metabolites in a biological sample, such as cell lysate or urine, leveraging the high mass accuracy and resolution of a Q-TOF system.
1. Sample Preparation:
- Precipitate proteins from the biofluid using cold methanol or acetonitrile (e.g., 1:3 sample:solvent ratio).
- Centrifuge to remove precipitated protein and transfer the clear supernatant to an LC vial.
- Create a quality control (QC) sample by pooling a small aliquot of every sample in the study batch.
2. LC-QTOF Method Setup:
- Chromatography: Utilize a UHPLC system with a reversed-phase column (e.g., ACQUITY UPLC BEH C18). Employ a water/acetonitrile gradient with 0.1% formic acid for positive ionization mode, or ammonium acetate or hydroxide for negative mode, over 10-20 minutes.
- MS Acquisition:
- TOF MS Scan: Set the quadrupole to RF-only mode to transmit all ions between m/z 50-1200. Acquire data in centroid mode with a scan rate of 1-10 spectra/second.
- Auto-MS/MS: Enable data-dependent acquisition (DDA). For each MS scan, select the top N (e.g., 5-10) most intense ions for subsequent MS/MS analysis. Set the quadrupole to isolate the selected precursor with a 1-4 m/z window. Fragment the ions in the collision cell using a collision energy ramp (e.g., 10-40 eV). The TOF analyzer then acquires the high-resolution product ion spectrum.
3. Data Processing and Compound Identification:
- Use dedicated software (e.g., T-ReX from Bruker, MarkerView, XCMS) for peak picking, alignment, and normalization [84].
- The software will generate a list of detected features, defined by their m/z and retention time.
- Filter features based on reproducibility in QC samples.
- Annotate significant features by querying accurate mass (± 5 ppm) against metabolomic databases (e.g., HMDB). Confirm identities by matching the acquired MS/MS fragmentation pattern with reference spectra from libraries [84].
Workflow Visualization: Untargeted Metabolomics on a Q-TOF
The following diagram illustrates the logical workflow and instrumental components involved in a standard untargeted metabolomics experiment using an LC-ESI-Q-TOF system.
Diagram 1: Untargeted Metabolomics LC-ESI-Q-TOF Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of mass spectrometric experiments, particularly in ESI-based research, requires a suite of high-purity reagents and consumables. The following table details key items and their functions.
Table 2: Essential Research Reagent Solutions for ESI-MS
| Item | Function / Application | Example Use Case |
|---|---|---|
| LC-MS Grade Solvents (Water, Methanol, Acetonitrile) | Mobile phase constituents; Minimize chemical noise and ion suppression for high-sensitivity detection. | Used in all LC-MS mobile phases and for sample reconstitution [83]. |
| Volatile Additives (Formic Acid, Ammonium Acetate) | Modulate pH for protonation/deprotonation in the ESI source; Improve chromatographic peak shape. | 0.1% Formic Acid for positive mode; 1-10 mM Ammonium Acetate for negative mode [85]. |
| Tuning & Calibration Solutions | Mass axis calibration and instrument performance verification. | Vendor-specific mixtures for quadrupole/ion trap; Sodium formate or Ultramark for TOF; Tunemix for FT-ICR. |
| Stable Isotope-Labeled Internal Standards | Normalize for matrix effects and variability in sample preparation and ionization efficiency. | Added to every sample and calibration standard in quantitative bioanalysis [85]. |
| Collision Gases (Nitrogen, Argon) | Inert gas for Collision-Induced Dissociation (CID) in tandem MS experiments. | Fills the collision cell in a QqQ or Q-TOF for fragmenting selected precursor ions [79] [81]. |
| Damping Gas (Helium) | Collisional cooling gas in ion trap instruments. | Improves trapping efficiency and mass resolution in QIT and FT-ICR cells [82] [83]. |
| Reference Mass Compounds | Real-time internal mass correction for high-accuracy measurements. | Leucine enkephalin infused via a LockSpray interface on Waters Q-TOF systems [85]. |
The landscape of mass analyzers offers a powerful yet diverse toolkit for the modern scientist engaged in ESI-based research. The choice between a quadrupole, ion trap, Q-TOF, or FT-ICR instrument is not a matter of selecting the "best" technology, but rather of aligning the instrument's core strengths with the specific analytical question at hand. As demonstrated in this guide, quadrupoles excel in robust, sensitive targeted quantification; ion traps provide deep insights into fragmentation pathways; Q-TOFs deliver a versatile balance of speed, resolution, and accuracy for untargeted discovery; and FT-ICR stands unparalleled for resolving the most complex mixtures with ultra-high precision. Understanding the fundamental principles, performance boundaries, and optimal application contexts of each analyzer, as summarized in the provided data tables and protocols, empowers researchers and drug development professionals to make informed decisions. This ensures that their analytical strategies are not only effective but also efficient, ultimately accelerating the pace of discovery and development in the life sciences.
Electrospray Ionization (ESI) revolutionized mass spectrometry by enabling the analysis of large, non-volatile biomolecules. As a soft ionization technique, it generates ions with minimal fragmentation by applying a high voltage to a liquid sample to create an aerosol of charged droplets, which undergo desolvation and Coulomb fission to produce gas-phase ions [1] [2]. The subsequent development of Secondary Electrospray Ionization (SESI) and Extractive Electrospray Ionization (EESI) represents a significant evolutionary trajectory aimed at overcoming fundamental limitations of conventional ESI, particularly for analyzing complex mixtures and trace compounds in challenging matrices without extensive sample preparation.
These derivative technologies have empowered real-time analysis of trace compounds residing in gases and aerosols, demonstrating remarkable potential across a wide spectrum of applications spanning disease diagnosis, drug detection, food safety, and environmental surveillance over the past three decades [86]. Concurrently, strides in deciphering the ionization mechanisms of SESI and EESI have spurred the creation of diverse ion source configurations characterized by enhanced sensitivity and diminished background noise, establishing their importance in modern analytical chemistry and drug development workflows.
Fundamental Ionization Mechanisms
From ESI to SESI and EESI: Core Principles
The fundamental innovation of SESI and EESI lies in their separation of the sample introduction process from the primary ionization event, which occurs in a confined region through the interaction of multiple plumes.
Secondary Electrospray Ionization (SESI) primarily focuses on the analysis of gaseous samples. In SESI, a primary electrospray generates charged solvent droplets and reagent ions. When gas-phase analyte molecules collide with this charged plume, they are ionized through mechanisms such as proton transfer or charge exchange, depending on the relative proton affinities and chemical properties of the reagents and analytes [86].
Extractive Electrospray Ionization (EESI) operates on a two-spray principle, where one spray generates charged solvent droplets via conventional electrospray, while a second, neutral spray introduces the sample as an aerosol. The two plumes intersect, and the collision between charged electrospray droplets and neutral sample droplets allows for the extraction of soluble components into the charged droplets. These droplets then undergo standard ESI processes—desolvation, droplet fission, and ion emission—to produce gas-phase ions of the extracted analytes [87] [88].
The following diagram illustrates the core ionization mechanism and instrumental setup of the EESI process:
Theoretical Models and Influencing Factors
The ionization efficiency in SESI and EESI is governed by several well-defined physical and chemical parameters. The final production of gas-phase ions is primarily explained by two major theories: the ion evaporation model (IEM), which suggests that as droplets reach a critical radius, field strength at the surface becomes sufficient to assist field desorption of solvated ions; and the charged residue model (CRM), which proposes that electrospray droplets undergo evaporation and fission cycles until progeny droplets contain approximately one analyte ion [1].
Key factors influencing ionization efficiency include:
Analyte Characteristics: Proton affinity, dipole moment, polarizability, and solubility significantly impact charge transfer and extraction efficiency [86]. Polar compounds with uneven charge distribution ionize more readily, while EESI can detect non-polar molecules using charged cations or anions to adsorb on polar groups [87].
Operational Parameters: Temperature, humidity, voltage, flow rates, and electrospray composition must be optimized for different applications [86]. For instance, slight decreases in sensitivity are observed with increasing absolute humidity for some ions [89].
Gas-Phase Extraction: The unique advantage of EESI lies in its gas-phase extraction process, which occurs away from the electrochemical circuit of the ESI needle, potentially preserving the original oxidation state of the analyte and minimizing sample degradation [88].
Technical Evolution and Instrumental Advances
Development of Source Configurations
The progression of SESI and EESI sources has seen continuous innovation to enhance performance characteristics:
Early SESI/EESI Designs: Initial configurations featured two sprayers (one charged, one neutral) positioned at specific angles (α, β, γ) and distances (a, b) from the mass spectrometer inlet to optimize collision efficiency and ion transmission [87].
Neutral-Desorption EESI (ND-EESI): This variant uses a neutral gas stream (e.g., nitrogen) to desorb analytes from surfaces like human skin or viscous materials, separating the sampling and ionization processes in time and space for remote analysis with minimal invasion [87] [90].
Internal EESI (iEESI): This approach directly introduces a charged solution into bulk samples, where the solvent diffuses and analytes move along the electric field gradient to form a stable electrospray plume at the mass spectrometer entrance [87].
Miniaturization and Specialization: Recent developments include nano-EESI systems operating at low flow rates (nL/min) for improved ionization efficiency and specialized configurations for high-throughput analysis [1].
Performance Comparison and Optimization
The table below summarizes key performance characteristics and their dependencies for EESI and SESI techniques:
Table 1: Performance Characteristics of SESI and EESI Techniques
| Performance Parameter | SESI Characteristics | EESI Characteristics | Key Dependencies |
|---|---|---|---|
| Detection Limit | ppt-ppb range for gases | 1-10 ng/m³ for aerosols [89] | Analyte properties, source design |
| Analysis Speed | Real-time (seconds) | Real-time (seconds) [87] | Flow rates, data acquisition |
| Matrix Effects | Moderate reduction | Significant reduction [88] | Extraction efficiency, spray stability |
| Dynamic Range | Linear over several orders | Linear response to mass [89] | Ionization mechanism, detector |
| Fragmentation | Minimal (soft ionization) | Minimal (soft ionization) [89] | Voltage settings, solvent composition |
Advances in understanding these parameters have enabled the creation of specialized sources with enhanced characteristics. For example, the EESI-TOF (Time-of-Flight) system provides online, near-molecular OA measurements at atmospherically relevant concentrations without analyte fragmentation, achieving detection limits of 1-10 ng/m³ in 5-second averaging times for typical atmospherically relevant compounds [89].
Experimental Protocols and Methodologies
Standard EESI-MS Analysis Protocol
Principle: EESI-MS enables direct analysis of complex mixtures without sample pretreatment by extracting analytes from a sample spray into a charged electrospray solvent plume during droplet collisions in the open air [87] [88].
Materials and Reagents:
- Mass spectrometer with API interface
- Two syringe pumps or HPLC pumps
- EESI source with two sprayers and adjustable positioning
- ESI solvent (e.g., methanol/water 1:1 v/v with 0.1% formic acid)
- Nitrogen nebulizing and drying gas
- Standard and sample solutions
Procedure:
- Source Setup: Mount two sprayers in the EESI source, aligning them to intersect at an optimal angle (typically 60-90°) approximately 5-10 mm from the mass spectrometer inlet.
- ESI Solvent Preparation: Prepare electrospray solution (methanol/water 1:1 v/v with 0.1% formic acid) to promote protonation.
- System Operation:
- Apply high voltage (3-5 kV) to the ESI sprayer
- Set ESI solvent flow rate to 3-5 μL/min
- Set sample flow rate to 3-5 μL/min (or use nebulizing gas)
- Optimize drying gas temperature and flow for desolvation
- Mass Spectrometer Operation:
- Set instrument to appropriate polarity (positive/negative ion mode)
- Optimize ion transfer voltages and collision energies
- Data Acquisition: Introduce samples and acquire mass spectra in full scan or tandem MS mode.
Applications: Direct analysis of urine, breath, foods, pharmaceuticals, and reaction monitoring [87] [90].
Neutral Desorption EESI for Surface Analysis
Principle: ND-EESI uses a jet of inert gas to desorb analytes from surfaces, which are then transferred to the electrospray plume for ionization, enabling minimal-contact sampling [87] [90].
Materials and Reagents:
- Neutral desorption gas (high-purity nitrogen)
- ND-EESI source with gas jet and transfer line
- Sample stage or holder
Procedure:
- Setup: Position the nitrogen jet 2-3 mm from the sample surface at a 30-45° angle.
- Gas Flow: Set nitrogen flow to 200-300 mL/min (velocity ~300 m/s).
- Sampling: Direct the gas jet at the sample area (~10 mm²) for 10-30 seconds.
- Transfer: Transport the desorbed analytes via transfer line to intersect with the electrospray plume.
- Analysis: Continue with standard EESI-MS analysis as described above.
Applications: Analysis of human skin, living organisms, fruits, cheese, and other surfaces without damage [90].
The following workflow diagram illustrates the key steps in the EESI experimental process:
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of SESI and EESI methodologies requires specific reagents and materials optimized for different analytical scenarios. The following table details key components of the research toolkit for these techniques:
Table 2: Essential Research Reagents and Materials for SESI/EESI Experiments
| Category | Specific Examples | Function/Purpose | Application Notes |
|---|---|---|---|
| ESI Solvents | Methanol/Water mixtures [1] | Primary electrospray solution | Adjustable ratio for optimal spray stability |
| Additives | Formic acid, Acetic acid (0.1%) [1] | Enhance conductivity and proton availability | Promotes [M+H]+ formation in positive mode |
| Additives | Ammonium acetate, Ammonia | Promote deprotonation for negative mode | Facilitates [M-H]- formation |
| Nebulizing Gases | Nitrogen (high purity) | Sample aerosolization and desolvation | Critical for ND-EESI and high-flow applications |
| Calibration Standards | Nicotine, Propanolol, Atrazine [87] [88] | System performance verification | Demonstrate wide linear dynamic range |
| Sample Introduction | Syringe pumps, LC pumps, Vaporizers | Controlled sample delivery to sprayer | Flow rates from nL/min to μL/min |
The selection of appropriate solvents and additives is particularly critical, as the polar compounds have an uneven charge distribution and large electron pair offset, making them easy to ionize, while non-polar molecules require charged cations or anions to adsorb on polar groups to generate molecular ions [87]. The unique configuration of EESI allows the use of different solvents for the electrospray and sample sprays, enabling optimization of ionization and extraction conditions independently.
Applications in Pharmaceutical Research and Drug Development
Real-Time Reaction Monitoring
EESI-MS enables direct, real-time monitoring of chemical reactions without sample pretreatment or extraction, providing insights into reaction kinetics, intermediates, and by-products. The technique uses two independent sprays—one for the sample solution and another generating charged solvent droplets—allowing direct trace detection of analytes in gas, liquid, or aerosol form without interference from complex matrices [87]. This capability is particularly valuable for:
- Reaction Optimization: Tracking reaction progress and identifying optimal termination points
- Intermediate Characterization: Detecting and identifying transient reaction intermediates
- By-product Identification: Revealing side products and decomposition pathways
- Catalysis Studies: Monitoring catalytic reactions and catalyst performance
Drug Metabolism and Pharmacokinetics
The high sensitivity and selectivity of SESI and EESI make them ideal for analyzing drugs and metabolites in complex biological matrices:
Breath Analysis: SESI-MS enables non-invasive detection of volatile biomarkers and drugs in exhaled breath, with applications in therapeutic drug monitoring and disease diagnosis [86] [90].
Biofluid Analysis: EESI-MS allows direct analysis of drugs and metabolites in urine, serum, and plasma without sample preparation, overcoming matrix effects that plague conventional ESI [88].
Tissue Imaging: Ambient ionization techniques like DESI (a related technique) enable mapping drug distribution in tissues, providing spatial resolution of drug and metabolite localization [91].
High-Throughput Screening and Biomarker Discovery
The rapid analysis capabilities of SESI and EESI support drug discovery workflows:
Metabolomics: EESI-MS facilitates rapid metabolic profiling for biomarker discovery and pathway analysis, with demonstrated applications in cancer research and metabolic disorders [87] [91].
Compound Screening: The technique's speed and sensitivity enable high-throughput screening of compound libraries against biological targets.
Quality Control: Direct analysis of pharmaceutical formulations and herbal medicines ensures product quality and detects contaminants or adulterants [87].
Comparative Analysis with Related Techniques
Advantages Over Conventional ESI and Other Ambient Methods
SESI and EESI offer distinct advantages that position them as valuable complements to existing ionization techniques:
Table 3: Comparison of ESI, SESI, EESI, and Related Ambient Ionization Techniques
| Technique | Sample Introduction | Key Advantages | Limitations |
|---|---|---|---|
| Conventional ESI | Liquid flow | Well-established, robust | Susceptible to matrix effects |
| SESI | Gas/vapor phase | Excellent for volatile analytes, real-time breath analysis | Limited to volatile/ semi-volatile species |
| EESI | Liquid, aerosol, gas | High matrix tolerance, minimal sample preparation | Optimization of two sprayers required |
| DESI | Solid surface | Spatial imaging capability | Surface topology effects |
| LAESI | Solid/biological | Direct tissue analysis, depth profiling | Requires mid-IR laser ablation |
The exceptional matrix tolerance of EESI stems from its gas-phase extraction process, which allows selective extraction of analytes from salt-rich solutions and increases analyte detection limits compared to direct ESI-MS analysis [88]. This makes it particularly valuable for direct analysis of biological fluids, environmental samples, and reaction mixtures without prior cleanup.
Technological Synergies and Hybrid Approaches
Recent developments have focused on combining the strengths of SESI/EESI with other analytical capabilities:
EESI-TOF: Coupling EESI with high-resolution time-of-flight mass spectrometry enables precise molecular formula assignments and untargeted analysis of complex mixtures [89].
EESI-Ion Mobility: Integration with ion mobility spectrometry provides additional separation dimension based on molecular shape and size.
Miniaturized Systems: Development of portable EESI and SESI sources for field analysis and point-of-care applications.
Future Perspectives and Concluding Remarks
The evolution of SESI and EESI represents a significant advancement in mass spectrometry ionization techniques, extending the capabilities of conventional ESI to address challenging analytical problems across pharmaceutical, clinical, and environmental domains. Future development trajectories are likely to focus on:
Enhanced Sensitivity and Throughput: Continued refinement of source designs, spray stabilization methods, and interface configurations to push detection limits and increase analysis speed.
Intelligent Automation: Implementation of machine learning algorithms for automated optimization of operational parameters and real-time data interpretation.
Single-Cell Analysis: Adaptation of EESI and SESI for single-cell metabolomics and proteomics, leveraging their minimal sample requirement and high sensitivity.
Standardization and Validation: Establishment of standardized protocols and validation guidelines to facilitate regulatory acceptance in pharmaceutical and clinical applications.
As these techniques continue to mature, their integration into mainstream analytical workflows will further expand their impact on drug discovery, personalized medicine, and environmental monitoring. The fundamental advantage of enabling direct, real-time analysis of complex samples without compromise continues to drive innovation in SESI and EESI technology, promising new capabilities for understanding molecular interactions in their native environments.
Conclusion
Electrospray Ionization has fundamentally transformed mass spectrometry, enabling the sensitive and accurate analysis of a vast range of biomolecules critical to clinical and pharmaceutical research. Its unique ability to transfer ions directly from solution to the gas phase underpins applications from high-throughput clinical screening to the study of delicate protein-ligand complexes. The future of ESI is directed toward greater miniaturization and automation, as seen in techniques like nano-DESI for spatial omics, and a deeper understanding of accelerated reactions in charged microdroplets for green synthesis and prebiotic chemistry. For biomedical researchers, mastering ESI principles, optimization strategies, and its contextual strengths compared to other ionization methods is essential for driving innovations in drug discovery, diagnostics, and personalized medicine.
References
- 1. Electrospray ionization [https://en.wikipedia.org/wiki/Electrospray_ionization]
- 2. Electrospray Ionisation Mass Spectrometry: Principles and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1853331/]
- 3. Electrospray Ionization [https://www.creative-proteomics.com/support/electrospray-ionization.htm]
- 4. Electrospray Ionization Mass Spectrometry [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Instrumentation_and_Analysis/Mass_Spectrometry/Mass_Spectrometers_(Instrumentation)/Electrospray_Ionization_Mass_Spectrometry]
- 5. Theoretical prediction of charged droplet evaporation and ... [https://www.sciencedirect.com/science/article/abs/pii/S1387380698141076]
- 6. Taylor cone [https://grokipedia.com/page/Taylor_cone]
- 7. Taylor cone [https://en.wikipedia.org/wiki/Taylor_cone]
- 8. Taylor Cone - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/taylor-cone]
- 9. Spontaneous Fission of Charged Water Nanodrops [https://pmc.ncbi.nlm.nih.gov/articles/PMC12147151/]
- 10. Current perspectives on supercharging reagents in ... [https://pubs.rsc.org/en/content/articlehtml/2021/ra/d1ra00745a]
- 11. The Generation of Multiply-charged Protein Anions from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7332380/]
- 12. Generation of multiply charged ions from homogeneous ... [https://www.sciencedirect.com/science/article/abs/pii/S1387380621000191]
- 13. Mass Analysis of Macro-molecular Analytes via Multiply- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7738406/]
- 14. Multiply-Charged Cation Attachment to Facilitate Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9670251/]
- 15. Section 2B. ESI-MS Data [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Analytical_Sciences_Digital_Library/In_Class_Activities/Biological_Mass_Spectrometry%3A_Proteomics/Instructors_Manual/Section_2%3A_Electrospray/Section_2B._ESI-MS_Data]
- 16. Exploring Charge-Detection Mass Spectrometry on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10568534/]
- 17. Charge Detection Mass Spectrometry: What's the “Big” Deal? [https://www.chromatographyonline.com/view/charge-detection-mass-spectrometry-what-s-the-big-deal-]
- 18. Mechanisms of ionization and of chemical reactions in ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d5sc04781a]
- 19. Mechanisms of ionization and of chemical reactions in ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04781a]
- 20. Imprint Desorption Electrospray Ionization Mass Spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12300929/]
- 21. Size-exclusion chromatography–electrospray-ionization ... [https://chemrxiv.org/engage/chemrxiv/article-details/67a330d46dde43c908a9adaa]
- 22. Tandem mass spectrometry in screening for inborn errors of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11882580/]
- 23. An Organic Chemist's Guide to Electrospray Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6384780/]
- 24. Tandem Mass Spectrometry Screening for Inborn Errors ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2021.631688/full]
- 25. Comparison of ESI– and APCI–LC–MS/MS methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC5762935/]
- 26. Visualizing Life With Ambient Mass Spectrometry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4297699/]
- 27. MAW: the reproducible Metabolome Annotation Workflow for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00695-y]
- 28. MALDI-ISD Mass Spectrometry Analysis of Hemoglobin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4496427/]
- 29. Identification of Human Hemoglobin Protein Variants Using ... [https://www.waters.com/nextgen/us/en/library/application-notes/2012/identification-human-hemoglobin-protein-variants-electrospray-ionization-electron-transfer-dissociation-mass-spectrometry.html?srsltid=AfmBOoozftd4SHSnkSr5LpxBCMyzSdGMZpu18wMVWDUGiX3DRHP9q4Wd]
- 30. Rapid Identification of Hemoglobin Variants by ... [https://pubmed.ncbi.nlm.nih.gov/11482884/]
- 31. A liquid chromatography-high-resolution mass ... [https://www.sciencedirect.com/science/article/pii/S2667145X25000021]
- 32. Dissociation Constant (KD) Simply Explained [https://fluidic.com/insight/dissociation-constant-kd-simply-explained-and-common-mistakes-to-avoid/]
- 33. Applications of ESI-MS in drug discovery: interrogation ... [https://www.nature.com/articles/nrd2083]
- 34. Application of Native ESI-MS to Characterize Interactions ... [https://www.sciencedirect.com/science/article/pii/S247255522206912X]
- 35. Underappreciated Chemical Interactions in Protein-Ligand ... [https://pubmed.ncbi.nlm.nih.gov/32016887/]
- 36. NMR-driven structure-based drug discovery by unveiling ... [https://www.nature.com/articles/s42004-025-01542-x]
- 37. A Browser-Based Tool for Assessing Accuracy of ... [https://pubmed.ncbi.nlm.nih.gov/40654087/]
- 38. Quantifying labile protein-ligand interactions using ... [https://pubmed.ncbi.nlm.nih.gov/20801056/]
- 39. Electrospray ionization (ESI) [https://nationalmaglab.org/user-facilities/icr/techniques/ionization-techniques/esi/]
- 40. The application of electrospray ionization mass ... [https://www.sciencedirect.com/science/article/pii/S0146638001001498]
- 41. the evolution of high-throughput mass spectrometry in ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d4np00026a]
- 42. HT SpaceM: A high-throughput and reproducible method ... [https://www.sciencedirect.com/science/article/pii/S0092867425009298]
- 43. High-throughput single cell metabolomics and cellular ... [https://pubmed.ncbi.nlm.nih.gov/35934357/]
- 44. How the Electrospray Ion Source market is expectd to grow ... [https://www.linkedin.com/pulse/how-electrospray-ion-source-market-expectd-grow-63-cagr-upcoming-otryc]
- 45. Direct Ionization Source Analysis Report 2025 [https://www.archivemarketresearch.com/reports/direct-ionization-source-803924]
- 46. Principles of Electrospray Ionization - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3134074/]
- 47. Optimization of the Electrospray Ionization Source with ... [https://www.spectroscopyonline.com/view/optimization-electrospray-ionization-esi-source-use-design-experiments-doe-approach-lc-ms-ms-determi]
- 48. 10 Great Tips for Electrospray Ionization LC–MS [https://www.chromatographyonline.com/view/the-lcgc-blog-10-great-tips-for-electrospray-ionization-lc-ms]
- 49. 6.3: Electrospray Ionization (ESI) Mass Spectrometry [https://phys.libretexts.org/Courses/University_of_California_Davis/Biophysics_241%3A_Membrane_Biology/06%3A_Experimental_Characterization_-_Mass_Spectrometry_and_Atomic_Force_Microscopy/6.03%3A_Electrospray_Ionization_(ESI)_Mass_Spectrometry]
- 50. Optimization of Electrospray Ionization by Statistical Design of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4972871/]
- 51. Principles of Electrospray Ionization [https://www.sciencedirect.com/science/article/pii/S1535947620301997]
- 52. An experimental design approach using response surface ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967309012643]
- 53. HPLC-Electrospray Ionization-Mass Spectrometry (LC-ESI-MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7874509/]
- 54. Electrospray Ionization (ESI) in LC-MS [https://www.metwarebio.com/electrospray-ionization-esi-lc-ms-analysis/]
- 55. Exploring the mechanism of salt-induced signal ... [https://pubmed.ncbi.nlm.nih.gov/25594702/]
- 56. Elucidating the Role of Ion Suppression in Secondary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10623576/]
- 57. A Corona Discharge Initiated Electrochemical Electrospray ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2770340/]
- 58. development and characterization of electrospray ionization ... [https://scholars.unh.edu/dissertation/2793/]
- 59. LC–MS Sensitivity: Practical Strategies to Boost Your ... [https://www.chromatographyonline.com/view/lc-ms-sensitivity-practical-strategies-boost-your-signal-and-lower-your-noise]
- 60. Nanopore ion sources deliver individual ions of amino ... [https://www.nature.com/articles/s41467-024-51455-x]
- 61. Enhancement of ionization efficiency of mass spectrometric ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267015011939]
- 62. Mobile Phase Optimization: A Critical Factor in HPLC [https://www.phenomenex.com/knowledge-center/hplc-knowledge-center/mobile-phase-in-chromatography?srsltid=AfmBOopk18fsNNqXsTK_6WP-pe7nMRchijeFstTHiOePmCE_XnwNUFya]
- 63. The critical role of mobile phase pH in the performance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8610006/]
- 64. Optimization of Electrospray Ionization Source Parameters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6297073/]
- 65. Cross validation of pharmacokinetic bioanalytical methods [https://www.sciencedirect.com/science/article/pii/S0731708524005272]
- 66. aspects of Mechanistic - the University of... electrospray ionization [https://research.rug.nl/en/publications/mechanistic-aspects-of-electrospray-ionization]
- 67. Some thoughts on electrospray ionization mechanisms [https://pubmed.ncbi.nlm.nih.gov/21719919/]
- 68. Method validation studies and an inter-laboratory cross ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6041428/]
- 69. Ionization Efficiency for Environmentally Relevant ... [https://www.chromatographyonline.com/view/ionization-efficiency-for-environmentally-relevant-compounds-using-atmospheric-pressure-photoionization-versus-electrospray-ionization]
- 70. Top 6 Ion Sources in Mass Spectrometry: EI, CI, ESI, APCI, ... [https://www.metwarebio.com/top-6-ion-sources-mass-spectrometry/]
- 71. 3.3: Atmospheric Pressure Ionization and Electrospray ... [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/An_Introduction_to_Mass_Spectrometry_(Van_Bramer)/03%3A_IONIZATION_TECHNIQUES/3.03%3A_Atmospheric_Pressure_Ionization_and_Electrospray_Ionization]
- 72. Mass Spectrometry Ionization Methods - Chemistry at Emory [https://chemistry.emory.edu/msc/tutorial/mass-spectrometry-ionization.html]
- 73. Electrospray ionization/atmospheric pressure photoionization ... [https://pubmed.ncbi.nlm.nih.gov/17428016/]
- 74. Comparison of atmospheric pressure photoionization ... [https://pubmed.ncbi.nlm.nih.gov/16478111/]
- 75. Comparison of electrospray ionization, atmospheric ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967312006127]
- 76. High resolution - ESI/APCI/APPI - FIA | Universidade de ... [https://www.usc.gal/en/services/area/research-infrastructures/services-and-equipment/mass-spectrometry-and-proteomics/high-resolution-esiapciappi-fia]
- 77. Straight to the Source: ESI vs APCI…. [https://www.microsaic.com/2020/01/13/straight-to-the-source-esi-vs-apci/]
- 78. Comparison of Electrospray Ionization and Atmospheric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4383307/]
- 79. Quadrupole mass analyzer [https://en.wikipedia.org/wiki/Quadrupole_mass_analyzer]
- 80. Quadrupole Mass Analyzer - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/quadrupole-mass-analyzer]
- 81. Quadrupole Mass Analyzer - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/quadrupole-mass-analyzer]
- 82. Mass Analyzers: An Overview of Several Designs and ... [https://www.spectroscopyonline.com/view/tutorial-mass-analyzers-overview-several-designs-and-their-applications-part-ii]
- 83. Ultra-High Mass Resolving Power, Mass Accuracy, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7031845/]
- 84. Quadrupole Time-of-Flight (QTOF) Mass Spectrometry [https://www.bruker.com/en/products-and-solutions/mass-spectrometry/qtof.html]
- 85. Q-Tof Premier Dynamic Range Enhancement and Accurate ... [https://www.waters.com/nextgen/us/en/library/application-notes/2006/q-tof-premier-dynamic-range-enhancement-and-accurate-mass-measurement-performance.html?srsltid=AfmBOor-WTaNj2RUk-JNAe6Kqd-vLP76Y2xtEQ35O12_1R2y5tGRsAkW]
- 86. The Evolution of Secondary /Extractive Electrospray : From... Ionization [https://pubmed.ncbi.nlm.nih.gov/40200688/]
- 87. Extractive electrospray ionization mass spectrometry for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9880169/]
- 88. Applications of extractive electrospray ionization (EESI) in ... [https://www.sciencedirect.com/science/article/abs/pii/S1387380615000603]
- 89. An extractive electrospray ionization time-of-flight mass ... [https://amt.copernicus.org/articles/12/4867/2019/]
- 90. Extractive electrospray ionization [https://en.wikipedia.org/wiki/Extractive_electrospray_ionization]
- 91. Mass Spectrometry Imaging [https://pubmed.ncbi.nlm.nih.gov/40674148/]