Specificity Testing in HPLC: A Comprehensive Guide to PDA and Mass Spectrometry Methods
This article provides a complete guide for researchers and pharmaceutical analysts on establishing method specificity using Photodiode Array (PDA) and Mass Spectrometry (MS) detection.
Specificity Testing in HPLC: A Comprehensive Guide to PDA and Mass Spectrometry Methods
Abstract
This article provides a complete guide for researchers and pharmaceutical analysts on establishing method specificity using Photodiode Array (PDA) and Mass Spectrometry (MS) detection. Covering foundational principles, practical methodologies, troubleshooting, and validation strategies, it addresses critical needs in analytical method development and compliance with ICH guidelines. The content explores the complementary strengths of PDA and MS for peak purity assessment, forced degradation studies, and impurity profiling, offering science-based best practices to ensure reliable and defensible analytical data for regulatory submissions.
Understanding Specificity: Core Principles and Regulatory Requirements
Defining Specificity in Analytical Method Validation
In the realm of analytical chemistry, particularly within pharmaceutical development and quality control, method validation provides documented evidence that a procedure is fit for its intended purpose. Specificity stands as a cornerstone validation parameter, demonstrating that an analytical method can accurately and unequivocally measure the analyte of interest in the presence of other components that may be expected to be present in the sample matrix [1]. This ensures the reliability of results, which is critical for drug efficacy, patient safety, and regulatory compliance.
The evaluation of specificity has evolved significantly with technological advancements. The International Council for Harmonisation (ICH) guidelines define it as the ability to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradation products, and matrix components [2]. Modern analytical techniques, notably High-Performance Liquid Chromatography coupled with Photodiode Array (HPLC-PDA) detection and Mass Spectrometry (MS), provide powerful orthogonal tools to confirm specificity, especially by verifying peak purity and identity [1] [3]. This article delineates the principles and practical protocols for establishing specificity within the framework of a broader thesis on analytical procedure validation.
The Critical Role of Specificity in Method Validation
Specificity, sometimes referred to as selectivity, ensures that a method's response is due solely to the target analyte. A lack of specificity can lead to inaccurate quantification, potentially masking instability (e.g., failure to detect degradation products) or overestimating potency, with serious consequences for drug quality [1]. The fundamental question specificity answers is: "Is the peak response due to a single component, free from co-elutions?" [1]
Specificity must be demonstrated for all types of analytical procedures:
- For Identification Tests: The method must be able to discriminate between compounds of closely related structure or to confirm the identity of an analyte via comparison with a known reference material [2].
- For Assay and Impurity Tests: It is crucial to demonstrate the resolution of the analyte from closely eluting compounds, which typically include the active ingredient, impurities, excipients, and degradation products [1]. This is achieved by spiking the sample with these potential interferents and proving that the assay result is unaffected.
Orthogonal Techniques for Specificity Assessment
Chromatographic separation is the first line of defense in achieving specificity. Parameters such as resolution, theoretical plate count (efficiency), and tailing factor are initial indicators [1]. However, adequate chromatographic separation alone does not conclusively prove peak homogeneity. Co-eluting compounds with similar retention times can go undetected without more sophisticated detection methods.
Photodiode Array (PDA) Detection
A PDA detector collects spectral data across a range of wavelengths for each data point across a chromatographic peak. This capability allows for:
- Peak Purity Assessment: By comparing spectra from the upslope, apex, and downslope of a peak, software algorithms can determine if the spectral profile remains consistent, indicating a pure peak, or changes, suggesting a co-eluting impurity [1] [2].
- Spectral Contrast: Software calculates the "purity angle" based on vector comparisons of spectra. A small angle suggests spectral homogeneity, while a larger angle indicates potential co-elution [2].
Limitations of PDA: Its effectiveness is limited if interfering compounds have no UV chromophores or very similar UV spectra. System noise and the relative concentration of the interferent can also impact its ability to distinguish minor co-elutions [1].
Mass Spectrometry (MS) Detection
Mass spectrometry provides a higher degree of specificity by separating and detecting ions based on their mass-to-charge ratio (m/z).
- Peak Purity and Identity: MS can provide unequivocal peak purity information, exact mass, and structural data. It is highly unlikely for two different compounds to co-elute and produce identical precursor and fragment ions [1] [4].
- Specific Detection in Complex Matrices: LC-MS/MS methods, operating in Multiple Reaction Monitoring (MRM) mode, are exceptionally specific and sensitive for quantifying analytes in complex biological matrices like plasma, as demonstrated in the quantification of potential anticancer drugs [4].
The combination of PDA and MS on a single HPLC instrument offers valuable orthogonal information. While PDA confirms spectral homogeneity, MS confirms identity and mass-based purity, creating a robust system to ensure no interferences are overlooked during method validation [1] [3].
The following workflow outlines the strategic application of these techniques for confirming specificity:
Experimental Protocols for Specificity Testing
The following protocols provide a detailed methodology for establishing the specificity of an HPLC method for a drug substance or product, incorporating both PDA and MS detection.
Protocol 1: Specificity and Forced Degradation Studies
This protocol is designed to demonstrate that the assay method is unaffected by the presence of impurities and degradation products [5].
- Materials: Drug substance, drug product (with excipients), known impurities, solvents (HPLC grade), acids (e.g., 0.1-1N HCl), bases (e.g., 0.1-1N NaOH), oxidants (e.g., 3% H₂O₂).
- Chromatographic System: HPLC system equipped with a PDA detector and a C18 column (e.g., 150-250 mm x 4.6 mm, 5 µm). The specific mobile phase and conditions should be optimized for the analyte [6] [5].
- Procedure:
- Prepare Solutions:
- Analyte Standard: Prepare a solution of the analyte at the target concentration.
- Stressed Samples: Subject the drug substance and product to various stress conditions:
- Acidic Hydrolysis: Treat with 0.1-1N HCl at room temperature or elevated temperature for a defined period (e.g., 24 h) [3] [5].
- Alkaline Hydrolysis: Treat with 0.1-1N NaOH at room temperature or elevated temperature for a defined period.
- Oxidative Degradation: Treat with 1-3% H₂O₂ at room temperature for a defined period [5].
- Thermal Degradation: Expose the solid drug to dry heat (e.g., 40-80°C) [3].
- Photolytic Degradation: Expose the drug to UV and/or visible light as per ICH guidelines [3].
- Placebo/Excipient Mixture: Prepare a solution containing all excipients at their respective concentrations in the drug product.
- Spiked Solution: Spike the analyte standard with known impurities and/or the placebo/excipient mixture.
- Chromatographic Analysis: Inject the above solutions into the HPLC system.
- Data Analysis:
- Examine the chromatograms for the resolution of the analyte peak from any degradation product peaks or excipient peaks.
- For the stressed samples, the peak purity of the main analyte should be assessed using the PDA detector. A pure peak confirms no co-elution with degradation products [5].
- Prepare Solutions:
Protocol 2: Peak Purity Assessment using HPLC-PDA
This protocol details the steps for performing peak purity analysis following a chromatographic run.
- Materials: The sample solutions from Protocol 1.
- Instrumentation: HPLC system with a PDA detector and associated software capable of peak purity analysis (e.g., LCsolution, Empower).
- Procedure:
- Set the PDA detector to acquire spectra across a suitable UV-Vis range (e.g., 200-400 nm) throughout the chromatographic run.
- Inject the sample and acquire data.
- In the data processing software, select the peak of interest (the analyte).
- Initiate the peak purity algorithm. The software will typically compare spectra from multiple points across the peak (e.g., upslope, apex, downslope).
- Interpret the results. The software provides a "purity angle" and a "purity threshold." A purity angle less than the purity threshold suggests a pure peak. Visually inspect the overlaid spectra for any deviations [1] [2].
Protocol 3: Confirmatory Analysis using LC-MS/MS
This protocol is used for definitive identification and to resolve any ambiguity from PDA results.
- Materials: The same sample solutions as in Protocol 1.
- Instrumentation: LC-MS/MS system with electrospray ionization (ESI).
- Procedure:
- Adjust the LC method if necessary to be compatible with MS detection (e.g., use volatile buffers like ammonium acetate or formate).
- For the analyte and any detected degradation products/impurities, establish the precursor ion ([M+H]⁺ or [M-H]⁻) and characteristic product ions via MS/MS fragmentation.
- Analyze the samples using a sensitive mode like MRM to monitor for the specific transitions of the analyte and potential degradants.
- Confirm the identity of the analyte peak by matching its retention time and MS/MS spectrum with that of a reference standard. The absence of other detectable ions at the analyte's retention time confirms peak purity from a mass spectrometric perspective [4] [3].
Data Presentation and Interpretation
The data generated from specificity experiments should be systematically evaluated and documented. Key aspects include resolution between critical pairs, peak purity results, and mass spectrometric confirmation.
Table 1: Specificity and Forced Degradation Results for a Hypothetical Drug Substance
| Sample Type | Stress Condition | Analyte Recovery (%) | Resolution from Closest Eluting Peak | PDA Peak Purity (Pass/Fail) | MS Confirmation of Degradant Identity |
|---|---|---|---|---|---|
| Unstressed Standard | - | 100.0 | - | Pass | - |
| Acid Degradation | 0.1N HCl, 24h, RT | 85.5 | 2.5 | Pass | DP-1 (Hydrolyzed product) |
| Base Degradation | 0.1N NaOH, 24h, RT | 78.2 | 3.1 | Pass | DP-2 (Hydrolyzed product) |
| Oxidative Degradation | 3% H₂O₂, 24h, RT | 92.1 | 4.0 | Pass | DP-3 (N-Oxide) |
| Drug Product (Placebo) | - | N/A | - | N/A | No interference detected |
Table 2: Key Reagent Solutions for Specificity Testing
| Research Reagent / Material | Function in Specificity Assessment |
|---|---|
| Drug Substance & Product | The primary materials to be analyzed and subjected to stress conditions to generate degradation products [5]. |
| Known Impurities | Used to spike the analyte to demonstrate resolution and the absence of interference in the assay [1]. |
| Placebo/Excipient Mixture | Represents the sample matrix without the active ingredient; used to prove the method's specificity towards the analyte in the presence of formulation components [5]. |
| Volatile Buffers (Ammonium Acetate/Formate) | Used in the mobile phase for LC-MS compatibility to prevent ion suppression and source contamination [3]. |
| Acids/Bases/Oxidants | Used in forced degradation studies to accelerate the formation of degradation products and demonstrate the stability-indicating property of the method [5]. |
Defining and demonstrating specificity is a non-negotiable requirement for any validated analytical method. While chromatographic parameters provide the initial evidence, the combined power of PDA and MS detection offers a comprehensive, orthogonal strategy to unequivocally prove that a method is specific. The experimental protocols outlined herein, encompassing forced degradation and peak purity assessment, provide a robust framework for researchers to generate defensible data that meets rigorous regulatory standards. Incorporating these practices ensures the development of reliable, stability-indicating methods that are crucial for the accurate assessment of drug identity, potency, and purity throughout its lifecycle.
Analytical procedure validation is a critical process in the pharmaceutical industry to ensure that analytical methods are suitable for their intended use. The International Council for Harmonisation (ICH) guideline Q2(R2) provides a framework for the validation of analytical procedures for drug substances and products [7]. This guideline outlines key validation characteristics and methodologies to demonstrate that an analytical procedure is appropriate for assessing the identity, quality, purity, and potency of pharmaceuticals.
Within the broader context of analytical development, specificity testing represents a fundamental validation parameter that demonstrates the ability to unequivocally assess the analyte in the presence of components that may be expected to be present, such as impurities, degradation products, and matrix components. The application of advanced detection techniques including Photodiode Array (PDA) detection and Mass Spectrometry (MS) has become increasingly critical for establishing method specificity, particularly for complex molecules and biological therapeutics.
The regulatory landscape for analytical method validation is primarily governed by ICH guidelines, with regional implementation by regulatory bodies including the U.S. Food and Drug Administration (FDA) and scientific standards established by the United States Pharmacopeia (USP). ICH Q2(R2) applies to analytical procedures used for the release and stability testing of commercial drug substances and products, both chemical and biological/biotechnological [7].
The FDA incorporates these international standards into its regulatory framework, with recent developments showing a trend toward modernizing requirements, such as the phased reduction of animal testing requirements for certain products while maintaining rigorous scientific standards [8]. Recent draft guidance from June 2025 on Q1 stability testing indicates the ongoing evolution of regulatory expectations [9].
Table 1: Core Regulatory Guidelines for Analytical Procedure Validation
| Regulatory Body | Guideline | Scope and Application | Current Status |
|---|---|---|---|
| International Council for Harmonisation (ICH) | Q2(R2) | Validation of analytical procedures for drug substances and products; includes definitions and methodology for validation characteristics [7]. | Active Scientific Guideline |
| U.S. Food and Drug Administration (FDA) | Adoption of ICH Q2(R2) | Enforcement of validation requirements for marketing applications; part of a broader framework including stability testing (Q1) [9]. | Implemented |
| U.S. Food and Drug Administration (FDA) | Various | Modernizing regulatory science, including updated approaches to testing and evidence generation [8]. | Ongoing Initiatives |
Specificity as a Validation Characteristic
Definition and Importance
Specificity is the validation parameter that unequivocally assesses the analyte in the presence of components that may be expected to be present. This includes typical impurities, degradation products, matrix components, and other relevant interfering substances. According to ICH Q2(R2), specificity demonstrations are required for identification tests, purity tests, and assay procedures [7].
In the context of a broader thesis on procedure for specificity testing, the combination of PDA and mass spectrometry detection provides complementary orthogonal data to establish method specificity comprehensively. PDA detection offers spectral purity information and peak homogeneity assessment, while mass spectrometry provides structural confirmation and definitive identity verification through mass-to-charge ratio detection.
Experimental Protocols for Specificity Demonstration
Protocol for Specificity Testing Using PDA Detection
Objective: To demonstrate method specificity using Photodiode Array (PDA) detection by confirming peak homogeneity and purity for the analyte in the presence of potential interferents.
Materials and Equipment:
- HPLC system equipped with PDA detector
- Reference standards of analyte and potential impurities/degradants
- Appropriate chromatographic column and mobile phase
- Sample preparation materials
Procedure:
- Prepare individual solutions of the analyte, known impurities, degradation products, and placebo components.
- Inject each solution separately into the chromatographic system and record retention times and spectral data.
- Inject a mixture containing all components to demonstrate resolution.
- For peak purity assessment, collect UV spectra across the analyte peak (at upslope, apex, and downslope positions).
- Analyze spectral homogeneity using the PDA software's purity algorithm or by visual comparison.
- Document resolution factors between the analyte and closest eluting potential interferent.
Acceptance Criteria: The method is considered specific if:
- Resolution between analyte and all potential interferents is ≥ 1.5
- Peak purity index meets acceptance criteria (typically ≥ 990)
- No significant co-elution observed
Protocol for Specificity Confirmation Using Mass Spectrometry
Objective: To provide orthogonal confirmation of specificity through mass spectrometric detection and structural characterization.
Materials and Equipment:
- LC-MS system with appropriate ionization source
- Reference standards
- Suitable chromatographic conditions compatible with MS detection
Procedure:
- Establish LC-MS conditions with appropriate ionization parameters.
- Inject analyte standard and obtain mass spectrum to confirm expected mass.
- Inject potential interferents individually to confirm distinct mass spectra.
- Perform forced degradation studies (acid/base, oxidative, thermal, photolytic stress).
- Analyze stressed samples to confirm separation of degradation products from main peak.
- Use extracted ion chromatograms (EIC) for specific masses to confirm no co-elution of species with different mass-to-charge ratios.
- For definitive identification, employ MS/MS fragmentation to characterize potential degradants.
Acceptance Criteria:
- Analyte shows expected mass with appropriate adduct formation
- No significant interference at same retention time with different mass
- Degradation products are resolved and identifiable
Specificity Assessment Workflow Using Orthogonal Techniques
Comprehensive Validation Characteristics
While specificity is critical for method reliability, ICH Q2(R2) defines multiple validation characteristics that must be evaluated based on the analytical procedure's intended use. The guideline provides detailed methodologies for deriving and evaluating various validation tests for each analytical procedure [7].
Table 2: Validation Characteristics as Defined in ICH Q2(R2)
| Validation Characteristic | Identification | Testing for Impurities | Assay | Methodology and Purpose |
|---|---|---|---|---|
| Specificity | Yes | Yes | Yes | Ability to assess analyte unequivocally in the presence of components that may be expected to be present [7]. |
| Accuracy | No | Yes | Yes | Closeness of agreement between the accepted reference value and the value found. |
| Precision (Repeatability, Intermediate Precision) | No | Yes | Yes | Closeness of agreement between a series of measurements. |
| Detection Limit | No | Yes | No | Lowest amount of analyte that can be detected, but not necessarily quantified. |
| Quantitation Limit | No | Yes | No | Lowest amount of analyte that can be quantitatively determined. |
| Linearity | No | Yes | Yes | Ability to obtain results directly proportional to analyte concentration. |
| Range | No | Yes | Yes | Interval between the upper and lower concentrations with suitable precision, accuracy, and linearity. |
| Robustness | No | Should be considered | No | Measurement of capacity to remain unaffected by small, deliberate variations in method parameters. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of specificity testing protocols requires specific reagents, reference materials, and instrumentation. The following table details essential components for conducting comprehensive specificity validation using PDA and mass spectrometry detection.
Table 3: Research Reagent Solutions for Specificity Testing
| Item/Category | Function/Application | Specification Considerations |
|---|---|---|
| Analytical Reference Standards | Provides benchmark for identity, purity, and quantitative analysis | Certified reference materials with documented purity and traceability |
| Forced Degradation Reagents | Generation of potential degradation products for specificity assessment | Includes acid (HCl), base (NaOH), oxidant (H₂O₂), and appropriate solvents |
| Chromatographic Columns | Separation of analyte from potential interferents | Multiple column chemistries (C18, phenyl, HILIC) for robustness assessment |
| MS-Grade Mobile Phase Additives | LC-MS compatibility and optimal ionization | Ammonium formate/acetate, volatile acids (formic, trifluoroacetic) |
| Sample Preparation Materials | Extraction and cleanup of analytical samples | Solvents, filters, solid-phase extraction cartridges, and containers |
Integrated Approach to Specificity Validation
Establishing comprehensive method specificity requires an integrated approach that leverages the complementary strengths of multiple analytical techniques. The workflow below illustrates the decision process for specificity confirmation using orthogonal techniques.
Decision Pathway for Specificity Confirmation
Regulatory Considerations and Compliance Strategy
Successful regulatory submission requires careful attention to the implementation of ICH Q2(R2) recommendations with consideration of region-specific requirements. The FDA's current approach emphasizes scientific justification and risk-based validation strategies, aligning with the principles of ICH Q2(R2) [7] [9].
When developing specificity protocols, consider these strategic approaches:
Early Engagement: Discuss novel approaches or complex method validation strategies with regulatory authorities through scientific advice procedures.
Comprehensive Documentation: Maintain detailed records of all specificity experiments, including raw data from both PDA and MS detection.
Orthogonal Verification: Employ multiple techniques to build a compelling case for method specificity, particularly for complex biological products.
Risk-Based Approach: Focus specificity testing efforts on the most probable and critical potential interferents based on the product's composition, manufacturing process, and degradation pathways.
The integration of advanced detection technologies including PDA and mass spectrometry with robust chromatographic separation provides a solid foundation for meeting regulatory expectations for analytical method specificity. This approach aligns with the FDA's emphasis on modernized, scientifically rigorous testing strategies [8] while complying with international standards for analytical validation [7].
The Critical Role of Specificity in Pharmaceutical Quality Control
In pharmaceutical quality control, specificity is the paramount parameter that ensures an analytical method can unequivocally assess the analyte of interest in the presence of other components that may be expected to be present, such as impurities, degradants, or matrix components [10]. This distinguishing characteristic is what separates a scientifically sound method from a mere test procedure. Without demonstrated specificity, analytical results lack the fundamental integrity required for making critical decisions regarding drug safety, efficacy, and quality. The validation parameter confirms that the method is truly measuring what it purports to measure, providing confidence that quality control tests will accurately detect variations in product quality.
The importance of specificity extends throughout the drug development lifecycle and into commercial manufacturing. For identity tests, specificity provides confirmation of molecular structure. For assay and impurity tests, it ensures accurate quantification without interference from closely related substances. Regulatory authorities including the FDA, EMA, and ICH have established rigorous requirements for demonstrating specificity, particularly through guidelines such as ICH Q2(R1) [10]. In today's regulatory environment, where complex molecules and sophisticated formulations are increasingly common, the ability to prove method specificity has become more challenging yet more critical than ever for pharmaceutical manufacturers seeking to maintain compliance and ensure patient safety.
Theoretical Foundations: Specificity in the Context of PDA and Mass Spectrometry
Fundamental Principles of Detection Techniques
Photo-Diode Array (PDA) Detection operates on the principle of ultraviolet-visible spectroscopy, where compounds absorb light at characteristic wavelengths. PDA detectors capture full spectra simultaneously, enabling peak purity assessment by comparing spectra across the peak. This capability makes PDA particularly valuable for detecting co-eluting compounds with different UV spectra, a common challenge in pharmaceutical analysis. However, a significant limitation of PDA arises when analyzing compounds with similar chromophores or those lacking strong UV-absorbing properties, as they may not provide sufficient discrimination [11].
Mass Spectrometry (MS) detection, particularly tandem mass spectrometry (MS/MS), provides specificity based on molecular mass and fragmentation patterns. In LC-MS/MS, the first mass analyzer selects the precursor ion (parent molecule), collision-induced dissociation fragments this ion, and the second mass analyzer monitors specific product ions. This two-stage mass filtering provides exceptional selectivity even in complex matrices. The Selected Reaction Monitoring (SRM) mode on triple quadrupole instruments offers superior performance for target compound quantification due to its high specificity and sensitivity [12].
Comparative Analysis of Specificity Capabilities
Table 1: Comparison of Specificity Characteristics Between PDA and MS Detection
| Characteristic | PDA Detection | MS Detection |
|---|---|---|
| Basis of Specificity | UV-Vis spectral characteristics | Mass-to-charge ratio and fragmentation patterns |
| Spectral Information | Full UV-Vis spectra (190-800 nm) | Mass spectra and fragmentation patterns |
| Peak Purity Assessment | Directly via spectral comparison | Indirectly via unique ion ratios and transitions |
| Sensitivity | Limited by molar absorptivity | Typically higher, especially for trace analysis |
| Matrix Effects | Susceptible to interfering chromophores | Susceptible to ion suppression/enhancement |
| Structural Information | Limited to chromophore characteristics | Extensive structural information via fragmentation |
| Ideal Applications | Routine analysis of known compounds with strong chromophores | Complex matrices, trace analysis, structural elucidation |
The orthogonal specificity provided by combining PDA and MS detection creates a powerful tool for comprehensive method characterization. While PDA confirms purity through spectral homogeneity, MS provides confirmation through mass-based identification. For regulated methods, this complementary approach offers robust scientific evidence for method specificity that withstands regulatory scrutiny.
Advanced Applications and Experimental Protocols
Specificity Challenge Studies: Protocol for Forced Degradation
Forced degradation studies represent the most comprehensive approach to demonstrating specificity by intentionally stressing drug substances and products to generate degradants that might co-elute with the main analyte under normal conditions.
Experimental Protocol: Forced Degradation Study
Sample Preparation:
- Prepare drug substance and drug product samples at appropriate concentrations (typically 1 mg/mL)
- Include stressed and unstressed controls
- For drug product, include placebo formulations without API
Stress Conditions:
- Acidic Hydrolysis: 0.1N HCl at room temperature or elevated temperature (e.g., 60°C) for various time periods
- Basic Hydrolysis: 0.1N NaOH at room temperature or elevated temperature for various time periods
- Oxidative Stress: 3% H₂O₂ at room temperature for various time periods
- Thermal Stress: Solid and solution states at elevated temperatures (e.g., 70°C)
- Photolytic Stress: Exposure to UV and visible light per ICH Q1B guidelines
Chromatographic Conditions:
- Column: ACE C18 (150 × 4.6 mm, 3 μm) or equivalent [11]
- Mobile Phase: Optimized for separation (e.g., acetonitrile:water [89:11, v/v] for triterpenoid acids) [11]
- Column Temperature: 20-35°C (optimized for critical separations) [11]
- Detection: Simultaneous PDA (190-400 nm) and MS detection
- Injection Volume: Typically 10-20 μL
Specificity Evaluation:
- Peak Purity: Assess via PDA spectral comparison across the peak (purity angle < purity threshold)
- Resolution: Ensure baseline separation between analyte and nearest degradant (R > 2.0)
- Identification: Use MS to identify major degradants via mass and fragmentation patterns
Case Study: Specificity Demonstration for Triterpenoid Analysis
Recent research on triterpenoid analysis in lingonberry extracts demonstrates the practical application of specificity protocols. The study developed and validated HPLC-PDA methods for determining 13 triterpenoids in complex plant matrices [11]. The methodology addressed the significant challenge of detecting triterpenoids with weak chromophores by employing detection at low wavelengths (205-210 nm) and optimizing mobile phase composition to enhance detection sensitivity while maintaining selectivity [11].
The validation demonstrated acceptable analytical specificity, confirming the method could distinguish between structurally similar triterpenoid classes including oleanane (oleanolic acid, β-amyrin), ursane (ursolic acid, α-amyrin), and lupane (betulinic acid, betulin) series [11]. The research emphasized that "characterization of triterpenoids can be carried out by a variety of chromatographic techniques, but the simultaneous determination of triterpenoids is rather challenging considering their similar structures and polarities, as well as the limitations of methodologies" [11], highlighting the critical importance of well-designed specificity studies.
Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Specificity Testing
| Reagent/Material | Function in Specificity Testing | Application Notes |
|---|---|---|
| Reference Standards | Provides authentic specimens for retention time and spectral comparison | Use highly purified characterized materials; include potential impurities and degradants |
| Forced Degradation Reagents (HCl, NaOH, H₂O₂) | Generates degradants for specificity challenge studies | Use high-purity reagents; prepare fresh solutions; include appropriate safety controls |
| HPLC-Grade Solvents (acetonitrile, methanol, water) | Mobile phase components for chromatographic separation | Low UV absorbance; minimal particulate matter; degas before use |
| Stationary Phases (C18, C8, phenyl) | Chromatographic separation media | Select based on analyte characteristics; various particle sizes and dimensions available |
| Mass Spectrometry Solvents (formic acid, ammonium acetate) | Mobile phase modifiers for MS compatibility | Volatile buffers preferred; concentration optimization critical for ionization efficiency |
| Internal Standards | Compensates for analytical variability in quantitative MS methods | Stable isotope-labeled analogs preferred; should mimic analyte behavior without interference [12] |
The selection of appropriate reagents and materials is critical for meaningful specificity demonstration. Internal standards deserve particular attention, as they "compensate for the variabilities in the analytical method, including sample preparation" and can be added "before the extraction – compensating for signal variation and the matrix effect plus extraction efficiency and extraction variability (recovery)" [12]. Properties of the internal standard should closely match those of the analyte, including extraction behavior, matrix effects, and ionization characteristics [12].
Workflow Visualization: Specificity Verification Protocol
Regulatory Considerations and Compliance Framework
Specificity demonstration is not merely a scientific exercise but a regulatory requirement embedded in current good manufacturing practices (cGMP). FDA inspections of pharmaceutical quality control laboratories comprehensively evaluate "specifications and analytical procedures" to ensure they are "suitable and, as applicable, in conformance with application commitments and compendial requirements" [13]. Regulatory scrutiny extends to examination of "chromatograms and spectra for evidence of impurities, poor technique, or lack of instrument calibration" [13].
During pre-approval inspections for NDAs/ANDAs, FDA inspection teams compare "the results of analyses submitted with results of analysis of other batches that may have been produced" and evaluate "methods and note any exceptions to the procedures or equipment actually used from those listed in the application" [13]. The agency specifically examines "raw laboratory data for tests performed on the test batches (biobatches and clinical batches)" and compares "this raw data to the data filed in the application" [13]. This level of scrutiny underscores the critical importance of robust, well-documented specificity studies in regulatory submissions.
The growing complexity of pharmaceutical molecules, including biologics, gene therapies, and personalized medicines, presents new challenges for specificity demonstration [14]. These advanced modalities require specialized validation approaches that address their unique characteristics, such as complex impurity profiles and sophisticated analytical techniques. The fundamental requirement for demonstrated specificity remains constant, though the methodologies continue to evolve with technological advancements.
Specificity stands as the cornerstone of reliable analytical methods in pharmaceutical quality control. The integration of orthogonal detection techniques, particularly PDA and mass spectrometry, provides comprehensive specificity verification that withstands both scientific and regulatory scrutiny. As the pharmaceutical landscape evolves with increasingly complex molecules and sophisticated formulations, the principles and protocols for specificity demonstration remain fundamental to ensuring drug quality, safety, and efficacy. The experimental approaches and regulatory framework outlined in this document provide a solid foundation for developing, validating, and maintaining specific analytical methods throughout the drug product lifecycle.
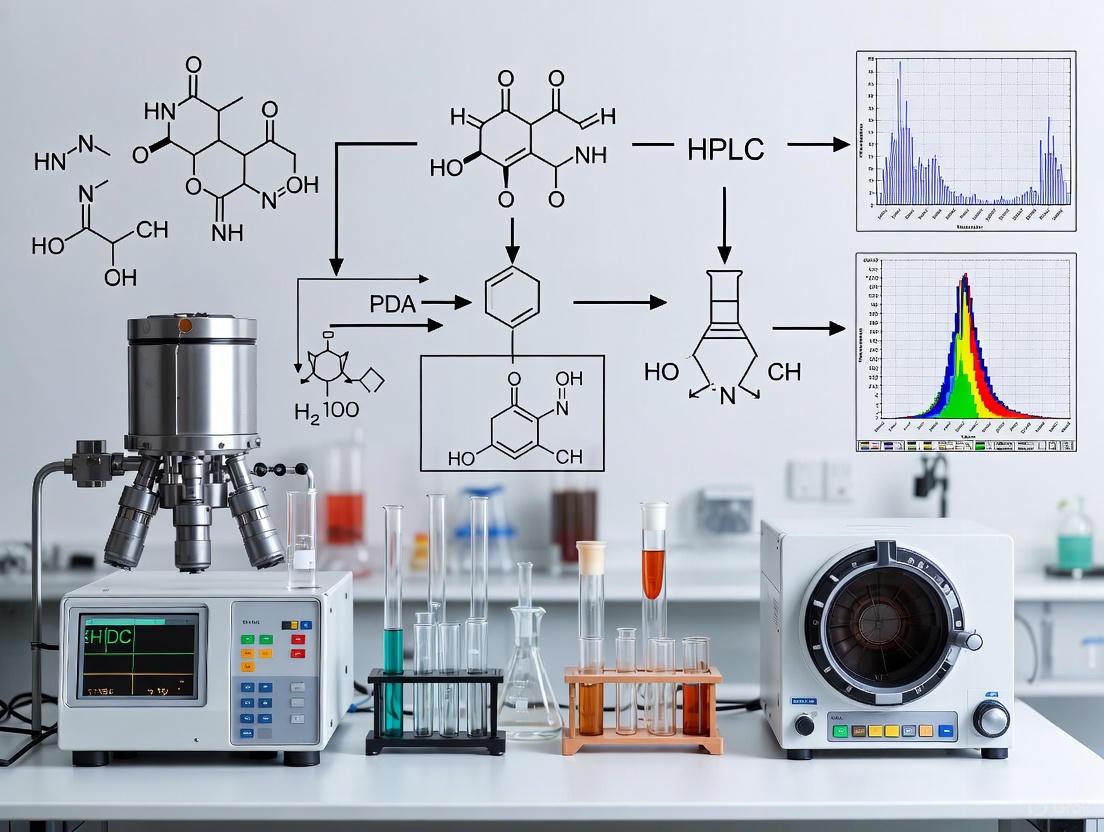
In pharmaceutical research and drug development, ensuring the specificity of an analytical method is paramount. Specificity confirms that the method can accurately and reliably measure the analyte of interest in the presence of other components, such as impurities, degradation products, or matrix elements. Photo-Diode Array (PDA) detection and Mass Spectrometric (MS) detection are two cornerstone technologies for this purpose, often employed in conjunction with liquid chromatography (LC). PDA detectors provide spectral data to confirm peak purity and identity, while MS detectors offer unparalleled specificity through mass-based identification and structural elucidation. This note details the fundamental principles, experimental protocols, and application of these technologies for specificity testing within a rigorous analytical framework.
Fundamental Principles
Photo-Diode Array (PDA) Detection
A PDA detector is a type of ultraviolet-visible (UV-Vis) spectrophotometer that simultaneously captures absorbance data across a range of wavelengths.
- Principle of Operation: When light from a broadband source passes through the sample flow cell, a holographic grating disperses the transmitted light onto an array of silicon diode detectors. Each diode corresponds to a specific wavelength, allowing for the immediate capture of the entire absorbance spectrum during an analysis [15] [16].
- Spectral Information: The primary data outputs are the chromatogram (absorbance at a specific wavelength vs. time) and the spectrum (absorbance vs. wavelength at any point in time). This enables peak purity assessment by comparing spectra across different points of a chromatographic peak. A pure peak will have a consistent, homogeneous spectrum. PDA detectors also facilitate compound identification by matching sample spectra with reference standards [3] [16].
- Advantages and Limitations: PDA is a robust, universal, and non-destructive detection method. However, its specificity can be limited when analyzing compounds with similar chromophores or in complex matrices where co-elution occurs.
Mass Spectrometry (MS) Detection
Mass spectrometry identifies molecules based on their mass-to-charge ratio (m/z), providing a higher degree of specificity and sensitivity.
- Ionization Source: The sample must be ionized before analysis. Common interfaces for LC-MS include Electrospray Ionization (ESI), which works well for a wide range of polar molecules, including proteins and metabolites. In ESI, a high voltage is applied to the liquid stream, creating a fine aerosol of charged droplets that desolvate to yield gas-phase ions [15] [3].
- Mass Analyzer: This component separates ions based on their
m/z. Triple quadrupole mass spectrometers are widely used for quantitative and targeted analysis. They consist of three quadrupoles in series: Q1 (mass selection), Q2 (collision-induced fragmentation), and Q3 (mass analysis of fragment ions). This setup enables highly specific Multiple Reaction Monitoring (MRM) experiments [15]. - Data Interpretation: The
m/zof the intact molecular ion (the precursor) provides the molecular weight. The fragmentation pattern (the product ions) serves as a unique fingerprint, allowing for structural confirmation and the identification of unknown impurities or degradation products [3].
Complementary Detection Mechanisms
The following diagram illustrates how PDA and MS detectors provide complementary information from a single liquid chromatography stream.
Experimental Protocols for Specificity Testing
A well-designed specificity test proves that the method can distinguish the analyte from all potential interferents.
Protocol 1: Specificity via Forced Degradation with LC-PDA and LC-MS/MS Analysis
This protocol outlines a stability-indicating method development as per ICH guidelines [15] [3].
1. Goal: To demonstrate that the LC-PDA-MS/MS method can separate and identify the active pharmaceutical ingredient (API) from its degradation products.
2. Materials and Reagents
- API reference standard
- Pharmaceutical formulation (e.g., tablet, suspension)
- LC-MS grade solvents: Acetonitrile, Water
- Acids (e.g., 0.1 M HCl) and Bases (e.g., 0.1 M NaOH)
- Oxidizing agent (e.g., 3% H₂O₂)
- Volatile buffers: Ammonium acetate, Ammonium formate
3. Instrumentation
- HPLC System: Binary pump, autosampler, thermostatted column compartment.
- Detectors: PDA detector and Triple Quadrupole Mass Spectrometer.
- Column: Reversed-phase C18 column (e.g., 100–150 mm x 4.6 mm, 2.7–5 µm particle size) [15] [3] [16].
- Software: Data acquisition and processing software for chromatography and mass spectrometry.
4. Procedure
- Step 1: Chromatographic Separation
- Mobile Phase: Prepare a mixture of 1 mM ammonium acetate buffer (pH ≈5.3) and acetonitrile (25:75, v/v) [15]. Filter through a 0.22 µm PVDF filter.
- Flow Rate: 0.5 mL/min [15].
- Column Temperature: 40 °C.
- Injection Volume: 1–20 µL [15] [16].
- Gradient/Isocratic: Use isocratic or gradient elution as needed for separation.
Step 2: Detection Parameters
Step 3: Forced Degradation Studies
- Acid/Base Degradation: Treat the API solution with 0.1 M HCl or 0.1 M NaOH at room temperature for 1-24 hours. Neutralize at the end of the stress period.
- Oxidative Degradation: Treat the API solution with 3% H₂O₂ at room temperature for 1-24 hours.
- Thermal and Photolytic Stress: Expose solid API to heat (e.g., 60 °C) and UV light.
Step 4: Analysis
- Inject separately prepared solutions of the unstressed API, stressed API, and blank.
- Record chromatograms and spectra using both PDA and MS detectors.
5. Data Interpretation
- PDA Analysis: Check for the appearance of new peaks in the stressed sample chromatograms. Use the software's peak purity algorithm to confirm that the API peak is spectrally homogeneous and not co-eluting with a degradation product.
- MS Analysis: Identify unknown degradation products by interpreting their precursor ion
m/zand fragmentation patterns. Compare these with the known fragmentation pathway of the API [3].
Protocol 2: Specificity in Biological Matrices using LC-MS/MS
This protocol is tailored for assessing specificity in complex biological samples like plasma [15].
1. Goal: To confirm that the method is specific for the analyte in the presence of endogenous matrix components.
2. Additional Materials
- Control (drug-free) human plasma.
- Protein precipitation reagents (e.g., Acetonitrile, Methanol).
3. Procedure
- Sample Preparation: Use protein precipitation. Add a volume of acetonitrile (e.g., 3:1 ratio) to the plasma sample, vortex mix, and centrifuge. Dilute the supernatant with water if needed [15].
- Chromatography: Optimize the LC method to separate the analyte from isobaric matrix interferences. A core-shell particle column can provide high efficiency [15].
- MS Detection: Use MRM mode for highest specificity. Monitor at least two MRM transitions per analyte.
- Analysis: Analyze at least six different sources of control plasma. Check for the absence of significant interfering peaks at the retention times of the analyte and internal standard.
4. Data Interpretation
- The method is considered specific if the peak area of any interference in control matrix is less than 20% of the lower limit of quantification (LLOQ) for the analyte.
Performance Data and Comparison
The quantitative performance of PDA and MS detectors differs significantly, influencing their application. The following table summarizes key characteristics based on validated methods.
Table 1: Comparative Analytical Performance of PDA and MS Detectors
| Parameter | PDA Detector Performance | MS/MS Detector Performance |
|---|---|---|
| Typical Linear Range | 1.40 – 55.84 ng/mL (for bulk analysis) [15] | 2.79 – 111.68 µg/mL (for bulk analysis) [15] |
| Detection Capability | Nanogram levels [15] | Picogram to nanogram levels [15] |
| Specificity Strength | Spectral homogeneity, identity via UV spectrum [3] | Molecular weight, fragmentation pattern, MRM transitions [15] [3] |
| Recovery in Plasma | 94.27% [15] | 98.20% [15] |
| Key Application | Peak purity, quantification of major components [3] [16] | Identification and quantification of impurities, metabolites, bioanalysis [15] [3] |
Table 2: Example Chromatographic Conditions for Specificity Testing
| Component | Condition 1 (GPB Analysis) [15] | Condition 2 (Rivaroxaban Analysis) [3] | Condition 3 (Antibiotics Analysis) [16] |
|---|---|---|---|
| Column | Ascentis Express F5 (100 x 4.6 mm, 2.7 µm) | Kinetex C18 (150 x 4.6 mm, 5 µm) | Shim-pack GIS C18 (250 x 4.6 mm, 5 µm) |
| Mobile Phase | 1 mM Ammonium Acetate:ACN (25:75) | 20 mM Ammonium Acetate:ACN (65:35) | 0.05M Oxalic Acid, ACN, Methanol (Gradient) |
| Flow Rate (mL/min) | 0.5 | 1.0 | 1.5 |
| Detection | PDA (200 nm) & MS/MS | PDA & MS/MS | PDA (330 nm) |
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials
| Item | Function / Application |
|---|---|
| Core-Shell Particle C18 Column | Provides high-efficiency chromatographic separation with low backpressure, ideal for resolving complex mixtures [15]. |
| Ammonium Acetate Buffer | A volatile buffer salt essential for MS compatibility; it does not leave residues that can foul the ion source [15] [3]. |
| LC-MS Grade Acetonitrile | High-purity solvent minimizes background noise and ion suppression in mass spectrometry [15]. |
| Analytical Reference Standards | Highly purified compounds used for method development, calibration, and positive identification of analytes. |
| Stable Isotope-Labeled Internal Standard | Corrects for variability in sample preparation and ionization efficiency in quantitative LC-MS/MS, improving accuracy and precision. |
The overall process for establishing method specificity integrates all the components and protocols described above, from sample preparation to final data interpretation as shown below.
In the development and validation of analytical methods for drug development, demonstrating specificity—the ability to accurately measure the analyte in the presence of potential interferents—is paramount. This requirement is a cornerstone of regulatory guidelines from the ICH (Q2(R1)). Liquid Chromatography (LC) coupled with Photodiode Array (PDA) detection and Mass Spectrometry (MS) provides a powerful orthogonal approach for specificity testing. The core performance parameters that underpin this testing are chromatographic resolution, detector selectivity, and spectral peak homogeneity. This document details the theoretical basis, experimental protocols, and practical data analysis for evaluating these critical parameters within the context of procedure for specificity testing, providing application notes for researchers and scientists in drug development.
Theoretical Foundations and Key Definitions
Resolution (Rₛ)
Chromatographic resolution quantitatively measures the separation between two analyte peaks. It is a function of column efficiency (number of theoretical plates, N), the retention factor (k), and the selectivity factor (α). A resolution value of Rₛ ≥ 2.0 is typically targeted for a robust separation of two closely eluting peaks, indicating complete baseline separation. For critical pair separations in pharmaceutical analysis, a minimum resolution of 1.5 is often considered acceptable.
Selectivity (α)
Selectivity, or the separation factor (α), describes the relative retention of two components on a given chromatographic system. It is calculated from the adjusted retention times and is independent of column efficiency. A selectivity value of α = 1 indicates no separation, whereas values greater than 1 indicate the potential for separation. Method development aims to maximize selectivity for critical pairs through manipulation of the mobile phase composition, pH, temperature, and stationary phase chemistry [17].
Peak Homogeneity
Peak homogeneity assessment is a crucial selectivity-evaluation tool in LC method development that determines whether a chromatographic peak originates from a single compound (homogeneous) or from co-eluting substances (heterogeneous). This is critically evaluated using PDA detection by comparing spectra across different segments of the peak [18]. The underlying principle is that spectra from a pure compound will be identical, while spectra from a peak containing multiple compounds will vary. Advanced liquid chromatography technologies, including low-adsorption hardware and novel separation modes like slalom chromatography, are being developed to tackle challenges related to non-specific adsorption, carryover, and inadequate selectivity, thereby improving resolution and robustness for large biomolecules [17].
Table 1: Key Performance Parameters for Specificity Testing
| Parameter | Definition | Calculation Formula | Acceptance Criterion | Primary Influence |
|---|---|---|---|---|
| Resolution (Rₛ) | Measure of separation between two peaks. | ( Rs = 2 \times \frac{t{R2} - t{R1}}{w{b1} + w_{b2}} ) | ( R_s \geq 1.5 ) (minimum) | Column efficiency, selectivity, retention |
| Selectivity (α) | Relative retention of two components. | ( \alpha = \frac{k2}{k1} = \frac{t{R2} - tM}{t{R1} - tM} ) | ( \alpha > 1 ) | Chemical nature of analyte, mobile/stationary phase |
| Peak Homogeneity | Purity of a chromatographic peak. | Spectral similarity factor (e.g., ( 1000 \times r^2 )) or alternative algorithms [18]. | Homogeneous profile (no significant spectral variation). | Specificity of detection, sample complexity |
Experimental Protocols for Parameter Assessment
Protocol for Determining Resolution and Selectivity
1. Scope and Application This protocol describes the procedure for determining the chromatographic resolution and selectivity factor between the analyte of interest and its closest eluting impurity, degradation product, or excipient.
2. Experimental Procedure
- Chromatographic System: Ultra-High-Performance Liquid Chromatography (UHPLC) or High-Performance Liquid Chromatography (HPLC) system.
- Column: A suitable reversed-phase C18 column (e.g., 100 mm x 2.1 mm, 1.7-2.6 µm particle size).
- Mobile Phase: As per the analytical method. For method development, a gradient elution is often used initially.
- Detection: PDA detector, scanning from 200 nm to 400 nm.
- Sample Preparation:
- Standard Solution: Prepare a solution of the analyte at the target concentration.
- System Suitability Solution: Prepare a mixture containing the analyte and all known impurities/degradation products at appropriate levels (e.g., 0.1-0.5% relative to the analyte).
- Injection: Inject the system suitability solution and record the chromatogram.
3. Data Analysis
- Identify the "critical pair" (the two least-resolved peaks of interest, typically the analyte and its closest eluting impurity).
- Measure the retention times ((t{R1}, t{R2})) and the peak widths at baseline ((w{b1}, w{b2})) for the critical pair.
- Calculate Resolution (Rₛ) and Selectivity (α) using the formulas provided in Table 1.
- Acceptance Criteria: The resolution between the analyte and all potential interferents should be ≥ 1.5.
Protocol for Assessing Peak Homogeneity with PDA Detection
1. Scope and Application This protocol assesses the spectral homogeneity of the analyte peak in the presence of placebo and stressed samples to demonstrate method specificity [18].
2. Experimental Procedure
- Follow the chromatographic conditions described in Section 3.1.
- Sample Set:
- Analyte Standard: Pure analyte.
- Placebo/Blank: Sample matrix without the analyte.
- Stressed Sample: Forced degradation samples (e.g., acid/base, oxidative, thermal, photolytic stress).
- Data Acquisition: Acquire full UV-Vis spectra (e.g., 200-400 nm) continuously throughout the elution of the analyte peak. Ensure a high spectral resolution and acquisition frequency (e.g., 10-20 spectra per second).
3. Data Analysis: Spectral Comparison Algorithms The following workflow, which can be implemented in software like Excel or automated within instrument software, is recommended for a robust assessment [18]:
- Spectral Acquisition and Normalization: Export all spectra acquired across the peak's elution profile. Normalize each spectrum to its maximum absorbance value to compare spectral shape independent of concentration.
- Pairwise Linear Regression: Perform linear regression between each possible pair of normalized spectra from the peak. For each comparison, record the slope, intercept, and correlation coefficient (r).
- Statistical Analysis: Calculate the mean and standard deviation for the resulting populations of slopes, intercepts, and correlation coefficients.
- Ellipsoid Volume (EV) Calculation: Visualize the data variability by calculating the volume of an ellipsoid in a 3D Cartesian space where the axes are the standard deviations of the slope, intercept, and correlation coefficient, and the center is their mean.
- Purity Metric Calculation: Transform the ellipsoid volume into a final Peak Homogeneity Value (PHV) using the formula: PHV = -log₁₀(EV). A higher PHV indicates greater spectral homogeneity.
Table 2: Key Research Reagent Solutions for Specificity Testing
| Reagent / Material | Function / Application | Example & Notes |
|---|---|---|
| UHPLC/PDA System | High-pressure fluid delivery, separation, and spectral acquisition. | Agilent 1290, Waters Acquity H-Class. Provides high-resolution separation and continuous spectral data. |
| RP Chromatography Column | Stationary phase for analyte separation. | Kinetex EVO C18, 100 mm × 2.1 mm, 2.6 µm [18]. Provides efficient separation. |
| Reference Standards | Identification and system suitability. | USP reference standards (e.g., carbamazepine, diazepam) [18]. |
| Forced Degradation Reagents | Generation of potential degradants for specificity validation. | 0.1M HCl, 0.1M NaOH, 3% H₂O₂, for acid, base, and oxidative stress, respectively. |
| Data Analysis Software | Processing of chromatographic and spectral data for purity assessment. | Instrument vendor software (e.g., Chemstation) or custom scripts in Microsoft Excel/R/Python. |
Advanced Orthogonal Confirmation with Mass Spectrometry
While PDA-based peak homogeneity is powerful, it has limitations, including the inability to detect co-eluting compounds with identical or highly similar UV spectra. Liquid Chromatography-Mass Spectrometry (LC-MS) provides an orthogonal and highly specific layer of confirmation.
Experimental Workflow:
- LC-MS Analysis: Analyze the specificity sample set (analyte, placebo, stressed samples) using LC coupled with a high-resolution mass spectrometer.
- Data Acquisition:
- Use Full Scan MS to detect all ionizable components.
- Use Data-Dependent MS/MS (ddMS²) or Data-Independent Acquisition (DIA) to fragment ions and obtain structural information.
- Data Interpretation:
- Extracted Ion Chromatograms (XIC): Generate XICs for the exact mass of the analyte and potential degradants. A single, symmetric peak in the XIC for the analyte supports homogeneity.
- Mass Spectral Purity: The mass spectrum at the peak apex should be dominated by the analyte's ion and its expected isotopes/adducts, with no significant extraneous ions.
- Dereplication Strategies: For complex mixtures, such as natural product extracts, use mutually supportive data including retention time, accurate mass, and MS/MS fragmentation patterns to differentiate known compounds from novel entities [19]. This strategy can rapidly identify 38 different cytochalasin compounds in a fungal extract, for example [19].
The following workflow diagram illustrates the integrated strategy for specificity testing using both PDA and MS:
Diagram 1: Integrated workflow for specificity testing using LC/PDA and LC-MS.
A robust procedure for specificity testing is foundational for reliable analytical methods in drug development. A systematic approach that combines the assessment of chromatographic resolution and selectivity with a rigorous, algorithm-driven evaluation of PDA-based peak homogeneity provides a strong framework. The orthogonal confirmation offered by mass spectrometry is indispensable for overcoming the inherent limitations of UV detection, ensuring that the method is truly specific for its intended analyte. By adhering to the detailed protocols and data analysis strategies outlined in this document, researchers can generate high-quality, defensible data that meets the stringent requirements of regulatory bodies.
Practical Implementation: PDA and MS Specificity Testing Protocols
In pharmaceutical analysis, demonstrating that a chromatographic method can accurately measure the target analyte without interference from impurities is a critical regulatory requirement. Peak purity analysis using Photodiode Array (PDA) detection provides a powerful tool for this purpose by determining spectral homogeneity across a chromatographic peak [20]. It is essential to understand that peak purity assessed by PDA is spectral purity, not absolute chemical purity; it indicates whether multiple components with different spectral characteristics are co-eluting [21] [20]. This application note details the theoretical principles, experimental protocols, and algorithmic assessments for reliable peak purity analysis within the framework of analytical method validation.
Theoretical Foundation of Spectral Comparison
The Vector-Based Model of Spectral Similarity
Peak purity assessment in most commercial software is founded on treating UV-Vis spectra as vectors in n-dimensional space, where n corresponds to the number of data points in the spectrum [21]. This model facilitates the quantitative measurement of spectral similarity.
- Spectral Contrast Angle (θ): The angle between two spectral vectors provides a robust metric for similarity. A smaller angle indicates more similar spectra [21].
- Cosine Calculation: The cosine of the angle θ between two mean-centered spectral vectors, a and b, is calculated as [21]: cos(θ) = (a • b) / (||a|| ||b||)
- Correlation Coefficient (r): An equivalent measure uses the correlation coefficient between the two spectra. For mean-centered vectors, the correlation coefficient is identical to the cosine of the spectral contrast angle [21].
Algorithmic Purity Assessment
Software algorithms compare multiple spectra extracted from different segments of a chromatographic peak—typically at the upslope, apex, and downslope—against a reference spectrum, often taken at the peak apex [21]. The core principle is that if all extracted, normalized spectra are identical (i.e., the spectral contrast angle is zero), the peak is considered "pure" from a spectral perspective [20]. A significant spectral difference suggests a potential co-elution of multiple compounds [21].
Table 1: Key Parameters in Spectral Comparison Algorithms
| Parameter | Description | Impact on Purity Assessment |
|---|---|---|
| Purity Angle | The largest spectral contrast angle found between any spectrum in the peak and the reference spectrum [21]. | A larger purity angle indicates greater spectral variance, suggesting potential impurity. |
| Purity Threshold | The angle, derived from system noise and spectral characteristics, above which a peak is considered impure [22]. | The peak is considered spectrally pure if the Purity Angle is less than the Purity Threshold [22]. |
| Spectral Contrast Angle | The angle between two spectral vectors in n-dimensional space [21]. | Quantifies the degree of similarity between any two spectra; a value of zero indicates identical shape. |
Experimental Protocol for PDA Data Acquisition
Proper data acquisition is foundational to obtaining meaningful peak purity results. Adherence to the following protocol ensures data of sufficient quality for algorithmic assessment.
Critical PDA Instrument Method Parameters
Table 2: Essential PDA Method Parameters for Peak Purity Analysis
| Parameter | Recommended Setting | Rationale |
|---|---|---|
| Wavelength Range | Start above the UV cutoff of the mobile phase; extend to cover all analyte absorbance maxima [20]. | Prevents detector saturation from mobile phase absorbance and ensures collection of relevant spectral data [20]. |
| Spectral Resolution | 1.2 nm [20]. | Provides optimal spectral detail for accurate comparison. |
| Sampling Rate | Sufficient to acquire 12-20 spectra across the narrowest peak of interest [20]. | Ensures adequate spectral sampling across the entire peak profile. |
| Peak Absorbance | Maintain maximum spectral absorbance (MaxPlot) below 1.0 AU [20]. | Minimizes photometric errors and spectral distortion that can falsely indicate impurity [20]. |
Step-by-Step Workflow
- Method Setup: In the PDA instrument method, configure the parameters as specified in Table 2. A resolution of 1.2 nm and an appropriate sampling rate are critical for high-fidelity data [20].
- System Preparation: Equilibrate the HPLC system with the starting mobile phase. Ensure the detector lamp has sufficient energy and a stable baseline.
- Sample Analysis: Inject the sample. For method validation, analyze stressed samples (e.g., exposed to acid, base, oxidizer, heat, or light) to force degradation and challenge the method's specificity [21].
- Data Collection: The software will collect a three-dimensional data matrix (Absorbance × Time × Wavelength) for the entire chromatographic run [23].
- Peak Purity Processing: In the processing method, enable the peak purity function. Set the "Active Peak Region" to less than 100% if the baseline is noisy to exclude spectra from the baseline region [22]. Initially, use the "AutoThreshold" feature to determine the purity threshold, then validate it with replicate injections of a standard [22].
Essential Research Reagent Solutions and Materials
The following table catalogues the key reagents, standards, and materials required for conducting validated peak purity studies.
Table 3: Key Research Reagents and Materials for Peak Purity Analysis
| Item | Function / Purpose | Notes for Use |
|---|---|---|
| High-Purity Reference Standard | Serves as the benchmark for spectral identity and purity; used to establish the purity threshold [22]. | Should be of the highest available chemical purity and well-characterized. |
| Stressed Samples | Samples subjected to stress conditions (acid, base, oxidative, thermal, photolytic) to generate potential degradants [21]. | Used during method development and validation to challenge the method's ability to detect co-eluting impurities. |
| Chromatography-Mobile Phase Solvents | High-purity HPLC-grade solvents and buffers for the mobile phase. | UV cutoff must be considered when setting the wavelength range to avoid background absorption [20]. |
| Placebo/Excipient Mixture | A mixture of all inactive components in a drug product formulation. | Used to demonstrate the absence of interference from excipients with the analyte peak (specificity) [1]. |
| Available Impurity Standards | Chemically synthesized or isolated impurities and degradants. | Used to positively identify impurities and confirm separation from the main peak [1]. |
Validation and Regulatory Considerations
Integrating peak purity assessment into the broader analytical method validation framework is essential for regulatory compliance.
Role in Method Validation
Peak purity analysis is a direct test of the specificity of an analytical method, one of the fundamental validation parameters defined by ICH guidelines [1] [21]. A validated, stability-indicating method must demonstrate its ability to measure the analyte unequivocally in the presence of potential impurities and degradants [21].
Complementary Use with Mass Spectrometry
While PDA is a powerful tool, its limitations must be acknowledged. Structurally similar compounds, such as impurities and degradants, often have highly similar UV spectra, making them difficult to distinguish [21]. Mass spectrometry (MS) provides orthogonal detection based on mass-to-charge ratio, offering a higher degree of certainty for peak identity and purity [1] [21]. Research has demonstrated that combining PDA and MS data can lead to superior results, such as perfect discrimination between genuine, generic, and counterfeit medicines, outperforming the use of either detector alone [24]. Therefore, for critical applications, the complementary use of PDA and MS is the preferred strategy [1] [24].
In the field of drug development, confirming the specificity of an analytical method is paramount to ensuring that measurements are accurate, reliable, and free from interference. Within the context of a broader thesis on procedure for specificity testing using Photodiode Array (PDA) and mass spectrometry research, this document details advanced protocols for assessing mass spectral purity and deconvoluting complex spectra. The concurrent use of PDA and Mass Spectrometry (MS) detectors provides a powerful orthogonal approach; where PDA can detect co-eluting peaks with different UV profiles, MS provides definitive identification based on mass-to-charge ratio ((m/z)) and fragmentation patterns [25].
A significant challenge in direct infusion mass spectrometry (DI-MS) and even liquid chromatography-mass spectrometry (LC-MS) is the prevalence of chimeric fragmentation spectra. These occur when multiple precursor ions with similar (m/z) are co-isolated and fragmented simultaneously, producing composite MS2 spectra that hinder unambiguous compound identification [26]. Spectral deconvolution techniques are therefore critical for isolating pure component spectra from these complex mixtures, thereby confirming method specificity. This article provides detailed application notes and protocols for employing these techniques, framed within the rigorous requirements of pharmaceutical development.
The Challenge of Chimeric Spectra and Principles of Deconvolution
In conventional DI-MS or LC-MS/MS workflows, the quadrupole mass analyzer uses an isolation window (often 1-2 (m/z) wide) to select precursors for fragmentation. In complex samples, this can lead to the simultaneous isolation of multiple isobaric or nearly isobaric compounds. Upon fragmentation, the resulting MS2 spectrum contains fragments from all co-isolated precursors, creating a chimeric spectrum that is difficult to interpret and can lead to misidentification [26].
DI-MS²: A Deconvolution Method
The DI-MS2 method has been developed as a robust solution to this problem. This technique modulates the intensity of precursors and their fragments by moving the quadrupole isolation window in small, discrete steps across a targeted (m/z) range [26]. The underlying principle is that an ion's transmission efficiency through the quadrupole depends on its position within the isolation window; ions closest to the center are transmitted most efficiently. As the isolation window shifts, the intensity of a given precursor ion (and consequently, all of its fragment ions) rises and falls in a characteristic modulation pattern. Ions originating from different precursors, with slightly different (m/z) values, will exhibit distinct modulation patterns. Deconvolution algorithms can then use these patterns to reconstruct pure, component-specific fragmentation spectra from the acquired chimeric spectra [26].
The following diagram illustrates the logical workflow of the DI-MS2 method and the subsequent deconvolution process.
Experimental Protocols
This section provides a detailed, step-by-step protocol for implementing the DI-MS2 deconvolution method to confirm mass spectral purity.
Protocol: DI-MS2 for Spectral Deconvolution
Objective: To deconvolute chimeric MS2 spectra and obtain pure fragmentation spectra for individual components in an isobaric mixture.
Materials and Reagents:
- Mass Spectrometer: A high-resolution instrument equipped with a quadrupole mass analyzer, such as a Linear Ion Trap-Orbitrap (LIT-Orbitrap) or a Quadrupole-Orbitrap (Q-Orbitrap) [26].
- Sample: A mixture of isobaric or nearly isobaric analytes. For example, a model system could contain two compounds with a nominal mass of 342 Da and a (m/z) difference of 0.006 for a challenging separation [26].
- Solvent: Appropriate MS-grade solvents for sample preparation (e.g., methanol, acetonitrile, water with 0.1% formic acid).
Method:
- Sample Preparation: Prepare a stock solution of the isobaric mixture in a suitable solvent. Dilute the stock solution to a concentration within the linear dynamic range of the mass spectrometer (e.g., 1-10 µM).
- Instrument Setup:
- Ion Source: Use an electrospray ionization (ESI) source.
- Ionization Mode: Set to positive or negative mode, as appropriate for the analytes.
- Direct Infusion: Load the sample into a syringe and infuse directly into the ion source at a constant flow rate (e.g., 3-5 µL/min).
- MS1 Acquisition:
- Perform a full MS1 scan to confirm the presence of the isobaric precursors and to define the target (m/z) range for the DI-MS2 experiment.
- DI-MS2 Method Configuration:
The key to success lies in the careful optimization of the following parameters, which are summarized in Table 1 for different instrument types.
- Isolation Window Width: Set the initial width. A narrower window (e.g., 1 (m/z)) improves selectivity but may reduce sensitivity [26].
- Step Size: Define the increment by which the isolation window will move between consecutive MS2 scans. A smaller step size (e.g., 0.1 (m/z)) provides higher resolution in the modulation pattern but increases total acquisition time [26].
- Mass Resolving Power: Set the resolving power of the high-resolution mass analyzer (e.g., Orbitrap). A higher resolution (e.g., 60,000-120,000) improves mass accuracy and separation of closely spaced fragments but lengthens the scan cycle [26].
- Collision Energy: Optimize the normalized collision energy (e.g., 25-35%) to ensure sufficient fragmentation without completely destroying the precursor ions.
- Automatic Gain Control (AGC) Target: Controls the number of ions accumulated. A higher value improves signal-to-noise but may increase scan time and the potential for space-charge effects [26].
- Number of Microscans: The number of scans averaged per spectrum. Increasing this improves signal-to-noise but prolongs acquisition time [26].
- Data Acquisition:
- Initiate the DI-MS2 method. The instrument will automatically acquire a series of MS2 spectra as the isolation window steps across the predefined (m/z) range.
- Data Analysis and Deconvolution:
- Software: Use specialized software capable of DI-MS2 deconvolution. The software will:
- Extract the intensity of every precursor and fragment ion across all acquired MS2 spectra.
- Correlate the intensity modulation patterns of fragment ions with those of the precursor ions.
- Group fragments that share the same modulation pattern as a specific precursor.
- Reconstruct a pure, component-specific MS2 spectrum for each precursor ion.
- Software: Use specialized software capable of DI-MS2 deconvolution. The software will:
The conceptual process of how intensity modulation enables deconvolution is illustrated below.
Troubleshooting:
- Poor Signal-to-Noise: Increase the AGC target or the number of microscans. Check sample concentration and ionization efficiency.
- Incomplete Deconvolution: Reduce the step size to better capture the intensity modulation. Consider using a narrower isolation window if the (m/z) difference between isobars is large enough.
- Excessive Acquisition Time: Reduce the number of microscans, use a larger step size, or narrow the target (m/z) range.
Performance Data and Instrument Comparison
The performance of the DI-MS2 method is influenced by the instrument platform and the chosen parameters. The following table summarizes key findings from a systematic evaluation on two high-resolution platforms.
Table 1: Impact of Instrument Type and Settings on DI-MS2 Performance [26]
| Parameter | LIT-Orbitrap | Q-Orbitrap | Optimization Guideline |
|---|---|---|---|
| Deconvolution Quality | High (Avg. similarity score: 0.98) | Variable (Avg. score: 0.56 to 0.96) | LIT-Orbitrap is more robust for complex mixtures. |
| Analysis Speed | Baseline | ~4x Faster | Q-Orbitrap offers superior throughput. |
| Optimal Isolation Window | 1 - 2 (m/z) | 1 - 2 (m/z) | Balance between sensitivity and selectivity. |
| Critical Step Size | < (m/z) difference of isobars | < (m/z) difference of isobars | Essential for resolving isobars with small (m/z) differences (e.g., 0.006). |
| Effect of (m/z) Difference | Consistently high scores | High scores for differences > 0.02; poor for differences ~0.006 | Q-Orbitrap may be less suited for extremely complex samples. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of specificity confirmation protocols requires not only the right instruments but also the right materials and reagents. The following table lists key solutions used in the featured experiments and the broader field.
Table 2: Essential Materials and Reagents for MS-Based Specificity Testing
| Item | Function / Application |
|---|---|
| Isobaric Compound Mixtures | Model systems for developing and validating deconvolution methods. Example: Mixtures with (m/z) differences as low as 0.006 [26]. |
| High-Purity Solvents (MS-Grade) | Ensure minimal background interference and stable electrospray ionization. Examples: Methanol, Acetonitrile, Water with 0.1% Formic Acid. |
| Biocompatible LC Systems | For analyzing sensitive biomolecules. Systems like the Waters Alliance iS Bio HPLC or Agilent Infinity III Bio LC Solutions use specialized materials to prevent analyte adsorption and maintain recovery [25]. |
| Advanced Mass Spectrometers | Instruments like the Sciex ZenoTOF 7600+ (with EAD fragmentation) and Bruker timsTOF Ultra 2 (with ion mobility) provide deeper structural insights and enhanced separation for specificity confirmation [25]. |
| Chromatography Data Systems (CDS) | Software for instrument control, data acquisition, and analysis. Modern CDS often integrate deconvolution algorithms and performance tracking (e.g., Sciex OS, Thermo Fisher Chromeleon) [25]. |
| Real-Time Spectral Deconvolution Software | Specialized software that mathematically deconvolutes overlapping spectra from detectors like DAD during an HPLC run, providing real-time purity assessment [25]. |
Forced degradation, or stress testing, is an essential component of pharmaceutical development that intentionally degrades drug substances and products under severe conditions to identify likely degradation products, elucidate degradation pathways, and establish the intrinsic stability of molecules [27]. These studies are fundamentally required to demonstrate the specificity of stability-indicating analytical methods, particularly when developing procedures for specificity testing using Photodiode Array (PDA) and mass spectrometry detection [15] [28]. Regulatory guidelines including ICH Q1A(R2) recommend but do not specify detailed protocols for forced degradation, leaving scientists to design scientifically justified conditions specific to their molecules [15] [27]. This application note provides detailed protocols and frameworks for designing, executing, and interpreting forced degradation studies within the context of modern analytical techniques.
Objectives and Regulatory Framework
Primary Objectives
Forced degradation studies serve multiple critical functions in drug development:
- Establish degradation pathways of drug substances and products [27]
- Demonstrate specificity of stability-indicating methods by separating degradants from the active pharmaceutical ingredient (API) and from each other [15] [28]
- Identify degradation products that may form during storage and facilitate structural elucidation [27]
- Provide insight into molecular stability to guide formulation development and packaging selection [27]
- Generate representative samples for developing and validating stability-indicating methods [27] [28]
Regulatory Context
While forced degradation studies are a scientific necessity during drug development, they are not formally part of the ongoing stability program [27]. Regulatory guidelines from ICH (Q1A(R2), Q1B, Q2(R1)) provide general principles but lack specific experimental details [15] [27]. The U.S. Food and Drug Administration (FDA) recommends stress testing should be performed on a single batch during Phase III development, though starting earlier in preclinical phases is highly encouraged to provide timely recommendations for manufacturing process improvements [27] [28].
Designing Stress Conditions
Core Stress Factors
A minimal set of stress factors should include hydrolytic (acid and base), thermal, oxidative, and photolytic conditions [27] [28]. The selection of specific stress conditions should be consistent with the product's decomposition behavior under normal manufacturing, storage, and use conditions [28].
Table 1: Recommended Stress Conditions for Forced Degradation Studies
| Stress Factor | Recommended Conditions | Typical Duration | Key Considerations |
|---|---|---|---|
| Acid Hydrolysis | 0.1 M HCl at 40-60°C [27] | 1-5 days [27] | Neutralize after stress; avoid over-degradation |
| Base Hydrolysis | 0.1 M NaOH at 40-60°C [27] | 1-5 days [27] | Neutralize after stress; avoid over-degradation |
| Oxidative Stress | 3% H₂O₂ at 25-60°C [27] | 1-5 days [27] | Higher temperatures may accelerate decomposition |
| Thermal Stress | 60-80°C [27] | 1-5 days [27] | Solid state: evaluate physical changes |
| Photolytic Stress | Exposure per ICH Q1B [27] | 1-5 days [27] | Minimum 1.2 million lux hours visible and 200-watt hours/m² UV |
Optimization Strategy
The optimal degradation range is generally considered to be 5-20% of the main peak, with approximately 10% degradation often providing sufficient challenge for analytical method validation [27] [28]. However, for biological products with multiple degradation pathways, generating multiple variants even at higher concentrations may be beneficial [28].
Studies should begin with moderate conditions (e.g., 40°C for hydrolysis) and multiple time points (1, 3, 5 days) to monitor degradation progression [27]. This approach helps distinguish primary degradants from secondary degradation products and provides better understanding of degradation kinetics [27].
Figure 1: Forced Degradation Study Workflow
Analytical Method Considerations
Specificity Demonstration
The forced degradation samples challenge the specificity of analytical methods, ensuring accurate quantification of the active ingredient and reliable detection of degradants [1]. Specificity should be demonstrated by showing separation of the API from known and potential impurities and degradants [1]. For chromatographic methods, this is typically shown through resolution between closely eluting peaks [1].
Integration of PDA and Mass Spectrometry
Modern forced degradation studies benefit from orthogonal detection techniques:
PDA Detection: Enables peak purity assessment by collecting spectra across a range of wavelengths at each data point throughout the peak [1]. This helps confirm that a peak's response is due to a single component with no co-elutions [1].
Mass Spectrometry: Provides unequivocal peak purity information, exact mass, and structural data, overcoming limitations of PDA detection when spectra are similar or relative concentrations low [1]. LC-MS-IT-TOF (liquid chromatography with ion-trap and time-of-flight mass spectrometer) can characterize novel degradation products and suggest formation mechanisms [15].
The combination of both PDA and MS on a single HPLC instrument provides valuable orthogonal information to ensure interferences are not overlooked during method validation [1].
Experimental Protocols
Comprehensive Forced Degradation Protocol
Materials and Equipment
- Drug substance (API)
- 0.1 M HCl and 0.1 M NaOH solutions
- 3% hydrogen peroxide (H₂O₂)
- LC-MS grade acetonitrile and water
- Ammonium acetate or other suitable buffer salts
- Thermostatically controlled oven or water bath
- Photostability chamber meeting ICH Q1B requirements
- HPLC system with PDA and MS detectors
- Core-shell particle column (e.g., Ascentis Express F5, 2.7 μm, 100 × 4.6 mm) or equivalent [15]
Sample Preparation
- Prepare stock solution of the drug substance at approximately 1 mg/mL in suitable solvent [27]
- For solution state stresses, use this concentration directly
- For solid state stresses, use the pure API
Stress Procedures
Table 2: Detailed Stress Testing Protocol
| Stress Condition | Procedure | Termination Method |
|---|---|---|
| Acid Hydrolysis | Mix 1 mL API solution with 1 mL 0.1 M HCl, incubate at 60°C [27] | Neutralize with 0.1 M NaOH after specified time |
| Base Hydrolysis | Mix 1 mL API solution with 1 mL 0.1 M NaOH, incubate at 60°C [27] | Neutralize with 0.1 M HCl after specified time |
| Oxidation | Mix 1 mL API solution with 1 mL 3% H₂O₂, store at 25°C [27] | Dilute with mobile phase before analysis |
| Thermal (Solution) | Incubate API solution at 60°C protected from light [27] | Cool to room temperature |
| Thermal (Solid) | Expose solid API to 60°C in oven [27] | Dissolve in appropriate solvent |
| Photolysis | Expose solid API and solution to light per ICH Q1B [27] | Protect from further light exposure |
Analysis Parameters
- Use HPLC conditions optimized for the API separation
- Example: Mobile phase: 1 mM ammonium acetate buffer:acetonitrile (25:75, v/v, pH ≈5.30) [15]
- Flow rate: 0.5 mL/min, Column temperature: 40°C [15]
- Detection: PDA monitoring at appropriate wavelength (e.g., 200 nm for glycerol phenylbutyrate) and MS detection [15]
- Injection volume: 1.0 μL [15]
Method Validation Parameters
Forced degradation studies support method validation by challenging key parameters:
- Specificity: Demonstrate separation of degradants from API and from each other [1]
- Accuracy: Assess by spiking known impurities into API [1]
- Precision: Evaluate repeatability through multiple injections of stressed samples [1]
- Linearity: Establish for both API and degradants across relevant concentration ranges [1]
Figure 2: Analytical Technique Selection for Specificity Confirmation
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Forced Degradation Studies
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Acid Solutions | Acid hydrolysis studies | 0.1 M HCl [27] |
| Base Solutions | Base hydrolysis studies | 0.1 M NaOH [27] |
| Oxidizing Agents | Oxidative stress studies | 3% Hydrogen peroxide [27] |
| Buffer Salts | Mobile phase preparation | Ammonium acetate for LC-MS [15] |
| HPLC Columns | Chromatographic separation | Core-shell particle columns (e.g., Ascentis Express F5) [15] |
| LC-MS Grade Solvents | Mobile phase and sample preparation | Acetonitrile, water [15] |
| Photostability Chamber | Photolytic degradation studies | ICH Q1B compliant light sources [27] |
Data Interpretation and Reporting
Evaluating Results
- Assess degradation extent: 5-20% degradation is generally acceptable for method validation [27]
- Identify critical separations: Note any degradants that co-elute with API or with each other
- Evaluate mass balance: The sum of API and degradants should account for initial amount within reasonable limits (typically 95-105%)
- Characterize major degradants: Use MS data to identify major degradation products and propose structures
Documentation
Comprehensive documentation should include:
- Detailed experimental conditions (concentrations, temperatures, durations)
- Chromatograms from all stress conditions
- Peak purity reports from PDA detection
- MS spectra and proposed structures for degradants
- Assessment of method specificity for each stress condition
Properly designed forced degradation studies provide critical insights into drug molecule behavior and form the foundation for validated stability-indicating methods. By implementing the scientifically justified conditions and comprehensive protocols outlined in this application note, researchers can develop robust analytical methods that reliably monitor product stability throughout its lifecycle. The integration of PDA and mass spectrometry detection provides orthogonal confirmation of method specificity, ensuring accurate detection and quantification of degradation products that may impact drug product safety and efficacy.
Sample Preparation and Chromatographic Optimization for Specificity
In pharmaceutical analysis, method specificity is the ability to unequivocally assess the analyte in the presence of components that may be expected to be present, such as impurities, degradation products, or matrix components [29]. Within the broader context of analytical procedure validation, establishing specificity is a fundamental requirement that ensures the reliability of results for drug identity, potency, and purity assessments. This application note provides detailed protocols for developing and optimizing chromatographic methods to achieve specificity using Photodiode Array (PDA) and Mass Spectrometry (MS) detection. The integration of these orthogonal detection techniques provides a powerful framework for confirming analyte identity and detecting potential interferences [30] [31].
Modern approaches to natural product research and pharmaceutical analysis combine powerful metabolite profiling for compound annotation with targeted isolation of prioritized compounds [30]. The strategies outlined herein align with this paradigm, emphasizing how high-resolution chromatographic separations closely matched between analytical and preparative scales, coupled with advanced detection, form the cornerstone of specificity confirmation.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key materials and reagents essential for conducting specificity testing as described in the subsequent protocols.
Table 1: Essential Research Reagents and Materials for Specificity Testing
| Item | Function/Description | Application Context |
|---|---|---|
| XBridge BEH C18 Column [31] | Reversed-phase column with 5 µm particles; provides robust separation for a wide range of analytes. | Method development and robustness testing for specificity. |
| Chloroacetic Acid [31] | Mobile phase additive; modifies pH to control ionization and improve peak shape for acidic/basic compounds. | Optimization of chromatographic selectivity. |
| Alliance iS HPLC System with PDA [31] | Enables precise optimization of detector parameters (slit width, resolution) to enhance sensitivity and spectral fidelity. | Peak purity assessment and spectral comparison. |
| High-Resolution Mass Spectrometer [32] [30] | Provides accurate mass and fragmentation data for definitive compound identification and detection of co-eluting species. | Structural confirmation and impurity identification. |
| LCGC Certified Clear Glass Vials [31] | Ensure sample integrity and prevent extractable/leachable interference during analysis. | Reliable sample introduction for both PDA and MS detection. |
Sample Preparation Strategies for Specificity
Proper sample preparation is a critical first step in ensuring method specificity, as it removes potential interferents from the sample matrix.
Protocol: Sample Preparation for Ibuprofen Impurity Analysis
This protocol is adapted from a validated United States Pharmacopeia (USP) method for organic impurities [31].
- Materials: Ibuprofen active pharmaceutical ingredient (API), mobile phase (4g/L Chloroacetic Acid in 40:60 water:acetonitrile, pH 3.0), LC-certified glass vials.
- Procedure:
- Weigh an appropriate amount of ibuprofen API.
- Serially dilute the sample using the mobile phase as the diluent.
- Prepare a final sensitivity solution at a concentration of 0.005 mg/mL (5 ppm).
- Transfer the prepared solution to a certified glass vial for analysis.
- Rationale: Using the mobile phase for dilution minimizes baseline disturbances and ensures compatibility with the chromatographic conditions. The low concentration of the sensitivity solution is designed to challenge the method's ability to detect and resolve minor impurities in the presence of the main analyte.
Chromatographic Optimization for Enhanced Specificity
Chromatographic separation is the primary line of defense in achieving specificity. The goal is to resolve the analyte peak from all other potential components.
Workflow for Specificity Method Development
The following diagram illustrates the logical progression from initial setup to a specific analytical method.
Protocol: Optimizing PDA Detector Parameters for Sensitivity and Spectral Specificity
While default detector settings are a good starting point, optimizing them is crucial for detecting low-level impurities and obtaining high-fidelity spectra for purity assessment [31]. This protocol uses the Alliance iS HPLC System with PDA Detector as a model.
Initial LC Conditions:
- Column: XBridge BEH C18, 250 x 4.6 mm; 5 µm
- Mobile Phase: 4g/L Chloroacetic Acid in 40:60 water:acetonitrile, pH 3.0 (isocratic)
- Flow Rate: 2.0 mL/min
- Injection Volume: 10.0 µL
- Detection: PDA, 254 nm [31]
Parameter Optimization Sequence: Conduct the following optimization in sequence, using the USP signal-to-noise (S/N) ratio of the target analyte peak as the key metric.
- Data Rate: Inject the sensitivity solution at data rates of 1, 2, 10, and 40 Hz. Select the rate that yields 25–50 data points across the narrowest peak and the best S/N. A rate of 2 Hz is often optimal for standard HPLC peaks [31].
- Filter Time Constant: With the data rate fixed, evaluate filter settings (No Filter, Fast, Normal, Slow). A slower time constant (e.g., Slow) often reduces baseline noise and improves S/N [31].
- Slit Width: Test slit widths (e.g., 35 µm, 50 µm, 150 µm). A narrower slit improves spectral resolution, while a wider slit can increase light throughput and sensitivity. Choose the best balance for your application [31].
- Resolution (Bandwidth): Evaluate resolution settings from 1 nm to 20 nm. Higher values (e.g., 8-16 nm) can improve S/N but decrease spectral resolution. For specificity requiring pure spectra, a lower setting may be preferable [31].
- Absorbance Compensation: Activate this feature using a wavelength range where the analyte does not absorb (e.g., 310–410 nm). This reduces non-wavelength dependent noise and can significantly improve the S/N ratio [31].
Expected Outcome: A systematic optimization of these parameters has been shown to produce a 7-fold increase in the S/N ratio compared to default settings, dramatically enhancing the ability to detect and characterize low-abundance impurities [31].
Table 2: Impact of PDA Detector Parameters on Signal-to-Noise Ratio
| Parameter | Default Setting | Optimized Setting | Effect on Sensitivity (S/N) | Consideration for Specificity |
|---|---|---|---|---|
| Data Rate [31] | 10 Hz | 2 Hz | Increased | Ensures sufficient data points for accurate peak integration and shape analysis. |
| Filter Time Constant [31] | Normal | Slow | Increased | Reduces high-frequency noise, improving the detection limit for minor impurities. |
| Slit Width [31] | 50 µm | 50 µm (situation-dependent) | Minimal change in study | A smaller slit width provides better spectral resolution for peak purity analysis. |
| Resolution [31] | 4 nm | 4 nm (situation-dependent) | Minimal change in study | A lower resolution setting preserves spectral detail for library matching. |
| Absorbance Compensation [31] | Off | On (310-410 nm) | Increased (1.5x) | Reduces baseline drift, leading to more reliable purity assessment. |
Orthogonal Detection: PDA and MS for Specificity Confirmation
The Specificity Confirmation Pathway
PDA and MS detectors provide complementary information for confirming specificity. The following diagram illustrates how data from these detectors is synthesized to prove a method is specific.
Protocol: Peak Purity Assessment using Photodiode Array Detection
PDA detection is the primary tool for assessing chromatographic peak purity within a single run.
- Acquisition: Acquire chromatographic data with a PDA detector across a suitable UV range (e.g., 200-400 nm). Ensure the peak of interest has a sufficient signal-to-noise ratio (e.g., S/N ≥ 10 as per USP criteria [31]).
- Analysis: For the peak in question, extract spectra from the upslope, apex, and downslope.
- Assessment: Overlay the extracted spectra. A pure peak will exhibit spectral homogeneity, meaning all overlaid spectra are identical. Any significant divergence (> 99.9% match threshold is common) indicates a co-eluting impurity, compromising specificity [31].
- Reporting: Software algorithms provide a purity angle and purity threshold. The peak is considered pure if the purity angle is less than the purity threshold.
Protocol: Confirmatory Analysis using Mass Spectrometry
MS detection provides definitive evidence of specificity by confirming analyte identity and detecting co-eluting species with different mass-to-charge ratios.
- Acquisition: Use an LC-MS system with high-resolution capabilities (e.g., UHPLC-HRMS). Employ data-dependent acquisition (DDA) to obtain both precursor (MS1) and fragment (MS/MS) ion data [30].
- Analysis:
- Extracted Ion Chromatograms (XICs): Generate XICs for the exact mass of the analyte and suspected impurities. Specificity is demonstrated if the chromatographic peak in the total ion chromatogram (TIC) is composed primarily of the XIC for the analyte.
- Spectral Fidelity: Check the MS1 spectrum at different points across the peak. The spectrum should be dominated by the ion signal of the analyte. The presence of other significant ions that change in relative abundance across the peak suggests a co-eluter.
- Structural Confirmation: The MS/MS spectrum should match a reference standard or a library entry, providing a second orthogonal identifier beyond retention time [30].
- Advanced Applications: For complex mixtures, such as natural product extracts, ion mobility spectrometry (IM-MS) can be coupled with MS to separate isomeric compounds that are chromatographically co-eluting but have different collision cross-sections, thereby enhancing specificity [32].
Establishing method specificity is a multi-faceted process that extends beyond achieving baseline chromatographic separation. This application note has detailed a comprehensive strategy integrating robust sample preparation, optimized chromatographic and detector parameters, and orthogonal detection with PDA and MS. By following the structured protocols for PDA optimization—fine-tuning data rate, filter constant, and absorbance compensation—analysts can significantly enhance sensitivity for impurity detection. The subsequent peak purity assessment and mass spectrometric confirmation provide a defensible, data-rich framework for proving a method is specific. This rigorous approach is essential for meeting regulatory standards and ensuring the safety and efficacy of pharmaceutical products.
In pharmaceutical analysis, ensuring the specificity of analytical methods is paramount to guarantee drug safety and efficacy. Peak purity assessment is a critical component of specificity testing, designed to determine whether a chromatographic peak corresponds to a single chemical entity or contains co-eluting impurities [21]. This application note details the application of photodiode array (PDA) detection and spectral similarity metrics—specifically purity angle and purity threshold—within the broader context of analytical procedure development for drug substances and products. These tools are essential for complying with ICH Q2(R1) validation requirements and developing stability-indicating methods that can detect and identify potential degradation products [15] [21].
Theoretical Foundations
Spectral Similarity and Vector Analysis
The fundamental principle underlying spectral peak purity assessment is the treatment of spectra as vectors in n-dimensional space, where n represents the number of data points in the spectrum [21].
- Spectral Contrast Angle: The angle (θ) between two spectral vectors quantifies their similarity, calculated as the cosine of θ [21].
- Correlation Coefficient: An alternative approach uses the correlation coefficient between two spectra, which is equivalent to the cosine of θ when vectors are mean-centered [21].
For two spectra represented as vectors a and b, the spectral similarity is calculated as:
[ \cos \theta = \frac{\mathbf{a} \cdot \mathbf{b}}{\|\mathbf{a}\|\|\mathbf{b}\|} = \frac{\sum{j=1}^{n} aj bj}{\sqrt{\sum{j=1}^{n} aj^2 \sum{j=1}^{n} b_j^2}} ]
After mean-centering the vectors, (\cos \theta) equals the correlation coefficient r [21].
Purity Angle and Purity Threshold
PDA-based peak purity assessment relies on comparing spectra across a chromatographic peak [33] [34].
- Purity Angle: A numerical value representing spectral variation across the peak. It is the average angle between each spectrum in the peak and the spectrum at the peak apex [33].
- Purity Threshold: A reference value derived from baseline noise, representing the maximum allowable spectral variation for a peak to be considered pure [33].
The relationship between these parameters determines peak purity:
- Pure Peak: Purity Angle < Purity Threshold → A single component is likely present [34].
- Impure Peak: Purity Angle > Purity Threshold → Co-elution of multiple components is likely [34].
Experimental Protocols
Peak Purity Assessment Using PDA Detection
This protocol describes systematic peak purity evaluation for specificity testing during analytical method development.
Materials and Equipment
Table 1: Essential Research Reagent Solutions and Materials
| Item | Function | Example Specifications |
|---|---|---|
| PDA Detector | Simultaneous multi-wavelength detection | Configured for 190-380 nm range [15] |
| Core-Shell Column | Chromatographic separation | Ascentis Express F5, 2.7 µm, 100 × 4.6 mm [15] |
| Ammonium Acetate | Mobile phase buffer | 1 mM, pH ≈5.30 [15] |
| Acetonitrile | Mobile phase organic modifier | LC-MS grade [15] |
| Trifluoroacetic Acid | Mobile phase modifier | Analytical reagent grade [15] |
| Forced Degradation Reagents | Sample stress studies | HCl, NaOH, H₂O₂ [15] |
Chromatographic Conditions
- Column: Ascentis Express F5 (2.7 µm, 100 × 4.6 mm) or equivalent [15]
- Mobile Phase: 1 mM ammonium acetate buffer:acetonitrile (25:75, v/v) [15]
- Flow Rate: 0.5 mL/min [15]
- Column Temperature: 40.0 ± 0.1°C [15]
- Injection Volume: 1.0 µL [15]
- Detection: PDA, 200 nm (for glycerol phenylbutyrate) [15]
Step-by-Step Procedure
System Preparation
- Filter mobile phase through 0.22 µm PVDF filter
- Degas mobile phase by sonication for 5 minutes
- Equilibrate system until stable baseline is achieved
Data Collection
Spectral Acquisition Across Peak
Data Processing
- Apply baseline correction between peak start and stop limits [21]
- Normalize spectra to account for concentration differences
- Compute spectral similarity between apex spectrum and all other spectra in the peak
Peak Purity Determination
- Calculate purity angle and purity threshold using instrument software
- Compare values: Purity Angle < Purity Threshold indicates pure peak [34]
- Visually inspect overlaid spectra for consistency across the peak
Complementary Mass Spectrometry Analysis
LC-MS/MS provides orthogonal confirmation of peak purity, particularly when PDA results are inconclusive.
Mass Spectrometry Conditions
- Ionization: ESI+ and ESI- with MRM [15]
- Mass Range: m/z 100-800 [15]
- Nebulizing Gas (N₂): 3.0 L/min [15]
- Drying Gas (N₂): 15 L/min [15]
- Heat Block Temperature: 450°C [15]
Procedure
- Analyze samples using identical chromatographic conditions with MS detection
- Monitor for multiple precursor-product ion transitions
- Examine extracted ion chromatograms for co-eluting species
- Compare mass spectra across the chromatographic peak
Figure 1: Workflow for PDA-based peak purity assessment and confirmation.
Data Interpretation and Analysis
Quantitative Data Analysis
Table 2: Peak Purity Interpretation Guidelines
| Purity Angle | Purity Threshold | Interpretation | Required Action |
|---|---|---|---|
| < 0.2 | Any value | Peak is pure; spectra are nearly identical [33] | None; method suitable for intended use |
| > 1.0 | > Purity Angle | No obvious co-elution; spectral differences may be noise-related [33] | Investigate method robustness |
| Any value | < Purity Angle | Co-elution likely; spectral differences exceed noise effect [33] | Method optimization required |
| > 1.0 | < Purity Angle | High probability of co-elution [33] | Significant method modification needed |
Case Study: Glycerol Phenylbutyrate Analysis
A recent study demonstrates the application of these principles to glycerol phenylbutyrate (GPB) analysis:
- Method Details: LC-PDA and LC-MS/MS methods were developed and validated according to ICH Q2(R1) [15]
- Linearity: 1.40–55.84 ng/mL for LC-PDA; 2.79–111.68 µg/mL for LC-MS/MS [15]
- Recovery: 94.27% (LC-PDA) and 98.20% (LC-MS/MS) in plasma samples [15]
- Forced Degradation: GPB was unstable under acid, alkali, and oxidative conditions, generating a novel degradation product [15]
Practical Considerations and Limitations
- Structural Similarity: Impurities and degradation products often have similar UV spectra, making differentiation challenging [21]
- Concentration Effects: High concentrations (>1 AU) can cause spectral distortion and false impurity flags [33]
- Uniform Co-elution: PDA cannot detect impurities with identical spectral shape and elution profile [33]
- Complementary Techniques: MS detection provides orthogonal confirmation when PDA results are ambiguous [15]
Figure 2: Spectral similarity assessment using vector analysis.
Purity angle, purity threshold, and spectral similarity metrics provide a robust framework for peak purity assessment in pharmaceutical analysis. When implemented according to the protocols outlined herein, these tools enable scientists to develop specific, stability-indicating methods that comply with regulatory requirements. The combination of PDA detection for initial assessment with MS confirmation for ambiguous cases represents a comprehensive approach to specificity testing in drug development.
Overcoming Challenges: False Results and Method Optimization
Identifying and Mitigating False Positive/Negative PDA Results
Pancreatic Ductal Adenocarcinoma (PDA) presents significant diagnostic challenges due to its insidious progression and the absence of reliable early detection methods. The diagnostic landscape is primarily plagued by the limitations of conventional biomarkers and imaging techniques, which frequently yield false positive and false negative results with profound clinical implications. Carbohydrate antigen 19-9 (CA19-9) remains the most widely used serum biomarker for PDA, yet it demonstrates limited sensitivity (79%) and specificity (82%) that severely restrict its utility for population screening [35]. This diagnostic inadequacy is compounded by the fact that over 50% of PDA cases exhibit no discernible abnormalities on pre-diagnostic contrast-enhanced computed tomography (CT), the current imaging standard [36]. These limitations create critical diagnostic windows where early, treatable tumors remain undetected, while false positives lead to unnecessary invasive procedures and patient anxiety.
The clinical consequences of diagnostic inaccuracy in PDA are substantial. False negative results directly contribute to delayed interventions, when tumors have typically progressed to advanced, inoperable stages. Patients diagnosed with metastatic PDA face a devastatingly short median survival of less than 12 months, whereas those detected at early, localized stages can achieve a 5-year survival rate exceeding 44% with appropriate treatment [35]. Conversely, false positive diagnoses trigger unnecessary psychological distress, financial burden, and potential harm from invasive diagnostic procedures like endoscopic ultrasound with fine-needle aspiration. This review comprehensively addresses the sources of diagnostic inaccuracy in PDA detection and presents advanced methodologies to enhance specificity and sensitivity through integrated biomarker panels, artificial intelligence-enhanced imaging, and mass spectrometry-based protein profiling.
Limitations of Conventional Biomarkers and Imaging
The high false negative rate in PDA diagnosis stems from multiple interconnected factors that current clinical protocols struggle to overcome. Conventional contrast-enhanced CT, while the diagnostic standard, relies on macroscopic tumor visualization and fails to detect subtle parenchymal changes associated with early tumorigenesis [36]. Retrospective analyses confirm that more than half of all PDA cases show no discernible abnormalities on pre-diagnostic imaging, creating a critical detection gap during the clinically actionable phase of disease progression [36]. This fundamental limitation is compounded by the rapid progression of PDA, which transitions from subclinical to advanced disease within an estimated 12-18 months—a timeframe that often outpaces the detection capabilities of conventional imaging protocols.
The diagnostic challenge extends beyond technological limitations to interpretative variability. Subtle imaging findings suggestive of early PDA, such as pancreatic duct cutoff or mild dilatation, are not only difficult to visualize but also lack specificity, as they frequently present in patients without malignancy [36]. This ambiguity contributes to both false positives and false negatives, with inter-reader agreement among radiologists remaining notoriously low (Cohen's kappa = 0.3) [36]. The anatomical position of the pancreas further complicates detection, as its retroperitoneal location and proximity to other structures can obscure early tumors. Additionally, the biological heterogeneity of PDA means that tumors do not always present with classical imaging features, leading to misinterpretation, especially in cases with atypical morphological characteristics or unusual growth patterns that deviate from expected radiographic presentations.
Analytical Factors in Biomarker Detection
Traditional single-marker approaches like CA19-9 demonstrate inherent limitations that contribute significantly to diagnostic inaccuracy. Beyond its modest sensitivity and specificity, CA19-9 levels can be elevated in various benign conditions including pancreatitis, obstructive jaundice, and biliary tract diseases, generating false positive results that complicate diagnostic interpretation [35]. Furthermore, approximately 5-10% of the population lacks the Lewis antigen required for CA19-9 expression, producing false negative results in these individuals regardless of their disease status [35]. This fundamental biological constraint highlights the inadequacy of relying on a single biomarker for definitive PDA diagnosis.
Pre-analytical and analytical variables introduce additional variability that impacts result accuracy. Sample collection methods, processing delays, and storage conditions can alter biomarker stability and detectability, particularly for protein markers and circulating tumor DNA (ctDNA) with short half-lives [37]. Immunoassay variability, including differences in antibody specificity, lot-to-lot reagent variation, and platform-dependent detection thresholds, further contributes to inconsistent results across laboratories [35]. Even with standardized protocols, biological factors such as hemolysis, lipemia, and heterophilic antibodies can interfere with assay performance, generating both false positive and false negative findings that may escape quality control measures without orthogonal verification methods.
Advanced Approaches for Enhanced Specificity and Sensitivity
Machine Learning-Enhanced Biomarker Panels
The integration of machine learning (ML) with multiplexed biomarker profiling represents a transformative approach to overcoming the limitations of single-marker testing. Recent research demonstrates that ML algorithms can significantly enhance diagnostic accuracy by identifying optimal biomarker combinations that capture the complex biological heterogeneity of PDA. In a comprehensive development and validation study, researchers employed multiple ML algorithms to analyze 47 serum protein biomarkers measured via Luminex bead-based immunoassays across 355 individuals (181 PDA patients, 174 healthy controls) [35]. The CatBoost algorithm emerged as the most effective, achieving remarkable diagnostic performance that substantially surpassed conventional CA19-9 testing.
Table 1: Performance Comparison of ML-Based Biomarker Panel vs. CA19-9 Alone
| Diagnostic Method | AUROC (All Stages) | AUROC (Early Stage) | Sensitivity | Specificity |
|---|---|---|---|---|
| CA19-9 Alone | 0.952 | 0.868 | 79% | 82% |
| ML Panel (CatBoost) | 0.992 | 0.976 | 95.5% | 90.3% |
| Validated ML Panel | 0.977 | 0.987 | N/R | N/R |
The ML-driven approach identified CA19-9, GDF15, and suPAR as the most influential biomarkers within the panel, with SHapley Additive exPlanations (SHAP) analysis quantifying their relative contributions to classification accuracy [35]. This multi-marker strategy demonstrated particular value in early-stage detection, where conventional methods struggle most, improving the AUROC from 0.868 with CA19-9 alone to 0.976 with the integrated panel [35]. The validation in an independent cohort of 130 individuals confirmed the robustness of this approach, with AUROC values of 0.977 for all disease stages and 0.987 specifically for early-stage PDA, highlighting its generalizability across diverse populations [35].
Artificial Intelligence-Augmented Imaging
Artificial intelligence has demonstrated remarkable potential to enhance the detection of pre-diagnostic PDA by identifying subtle imaging signatures imperceptible to human observation. The Radiomics-Based Early Detection Model (REDMOD) exemplifies this approach, leveraging deep learning to extract and analyze textural and structural alterations in normal-appearing pancreas tissue on CT scans obtained months to years before clinical diagnosis [36]. When applied to pre-diagnostic CTs with a median lead time of 398 days, REDMOD achieved an AUC of 0.98, significantly outperforming radiologist interpretation (AUC 0.66) and demonstrating particularly superior specificity (90.3% vs. approximately 82% for conventional reading) [36].
The AI model's exceptional performance stems from its capacity to detect microarchitectural remodeling that precedes macroscopic tumor formation. Ablation studies identified textural heterogeneity features from gray-level co-occurrence matrices (GLCM) as the most predictive elements, aligning with known biological changes during pancreatic carcinogenesis [36]. This capability allows for identification of at-risk individuals during the clinically actionable pre-diagnostic phase when conventional imaging appears normal. Additionally, AI-driven volumetric pancreas segmentation provides reproducible, quantitative assessments of pancreatic morphology, overcoming the inter-reader variability that plagues manual segmentation and establishes a foundation for longitudinal tracking of subtle parenchymal changes [36].
Mass Spectrometry-Based Protein Profiling
Mass spectrometry (MS) offers a powerful, unbiased platform for biomarker discovery and validation that transcends the limitations of antibody-dependent immunoassays. MS-based proteomic profiling enables simultaneous quantification of thousands of proteins from minimal sample volumes, providing unprecedented depth for discovering novel biomarker signatures with enhanced specificity for PDA [38]. The technology's particular strength lies in detecting low-abundance proteins, post-translational modifications, and subtle metabolic shifts that may signal early malignancy but remain undetectable by conventional methods.
Table 2: Key Research Reagent Solutions for MS-Based Biomarker Discovery
| Reagent/Category | Specific Examples | Function in PDA Research |
|---|---|---|
| Sample Preparation | Albumin/IgG depletion columns, Trypsin/Lys-C digestion kits, TMT/Isobaric tags | Reduces sample complexity, enables multiplexed quantification, improves detection of low-abundance biomarkers |
| Separation | C18 LC columns, High-pH reverse phase cartridges, Strong cation exchange resins | Fractionates complex peptide mixtures to reduce interference and increase proteome coverage |
| Mass Spectrometry | LC-MS/MS systems, TripleTOF, Orbitrap platforms, Q-TOF instruments | Provides high-resolution accurate mass measurements for protein identification and quantification |
| Data Analysis | MaxQuant, Skyline, PEAKS, Scaffold DIA software | Enables statistical analysis, biomarker quantification, and pathway analysis of proteomic data |
| Validation | Stable isotope-labeled internal standards, PRM/SRM assay kits, Quality control materials | Facilitates transition from discovery to targeted verification with high precision and reproducibility |
The MS workflow for PDA biomarker development follows a structured pathway from discovery to validation. Initial untargeted or "shotgun" proteomics comprehensively profiles proteins and metabolites differentially expressed between patient subgroups, employing liquid chromatography-tandem mass spectrometry (LC-MS/MS) and label-free or isobaric tagging quantification methods [38]. Bioinformatics pipelines then prioritize candidate biomarkers through statistical analysis and pathway enrichment, followed by rigorous validation using targeted MS techniques like Selected Reaction Monitoring (SRM) or Parallel Reaction Monitoring (PRM) [38]. These validated assays offer exceptional precision, reproducibility, and low limits of detection suitable for quantifying low-abundance biomarkers in complex biological matrices, ultimately producing clinically applicable tests with minimized false positive rates.
Experimental Protocols for Specificity Testing
Protocol 1: Multiplex Biomarker Panel Development with Machine Learning Integration
Objective: To develop and validate a multiplex serum protein biomarker panel with enhanced specificity for PDA detection using machine learning integration.
Materials and Reagents:
- Luminex xMAP bead-based immunoassay kits (Human Angiogenesis/Growth Factor Panels, Human Cancer/Metastasis Biomarker Panels)
- Luminex 200 system or comparable multiplex analyzer
- Serum samples from well-characterized cohorts (PDA patients and controls)
- Assay buffer, matrix solution, wash buffer
- Biotinylated detection antibodies, streptavidin-phycoerythrin
- Calibration standards and quality control materials
Methodology:
- Sample Preparation: Collect serum samples following standardized protocols. Process within 2 hours of collection, aliquot, and store at -80°C. Avoid freeze-thaw cycles.
- Biomarker Quantification:
- Prewet each well with 100µL wash buffer, incubate 10 minutes
- Add 25µL each of standard, quality control, and assay buffer to designated wells
- Add 25µL matrix solution followed by 25µL serum sample
- Add 25µL fluorescently labeled beads conjugated with target-specific antibodies
- Incubate overnight at 4°C on plate shaker
- Wash twice with 200µL wash buffer, add 25µL biotinylated detection antibodies
- Incubate 1 hour at room temperature, add 25µL streptavidin-phycoerythrin
- Incubate 30 minutes, wash twice, add 100µL sheath fluid
- Analyze on Luminex system using xPONENT software
- Data Preprocessing: Normalize raw fluorescence intensities using five-parameter logistic regression. Apply logarithmic transformation to normalize distributions of biomarker concentrations.
- Machine Learning Analysis:
- Implement multiple ML algorithms (CatBoost, XGBoost, Random Forest, SVM)
- Split dataset into training (80%) and testing (20%) subsets with stratification by age and gender
- Perform five-fold cross-validation to optimize hyperparameters
- Apply SHAP analysis to determine feature importance
- Select optimal biomarker combination based on diagnostic performance
- Validation: Test final model on independent validation cohort using predefined classification thresholds.
Quality Control: Include duplicate samples, internal standards, and blinded quality control samples in each run. Monitor inter-assay coefficient of variation (<15% for all biomarkers).
Protocol 2: AI-Augmented CT Image Analysis for Early PDA Detection
Objective: To implement an AI-based radiomics model for detecting subtle pancreatic changes predictive of PDA on conventional CT scans.
Materials and Software:
- Contrast-enhanced CT scans (arterial or pancreatic phase)
- Python 3.8+ with TensorFlow/PyTorch, scikit-learn, SimpleITK libraries
- Radiomics feature extraction platforms (PyRadiomics)
- High-performance computing workstation with GPU acceleration
- Annotated reference datasets for model training
Methodology:
- Image Acquisition and Preprocessing:
- Acquire CT scans with standardized protocol: 100-120 kV, 400-600 mA, slice thickness ≤3mm
- Reconstruct images with 1mm slice thickness for high-resolution analysis
- Apply noise reduction filters while preserving texture information
- Pancreas Segmentation:
- Implement automated deep learning segmentation (U-Net or nnU-Net architecture)
- Manual verification and correction by expert radiologists
- Extract whole-pancreas volume, excluding surrounding vessels and structures
- Radiomic Feature Extraction:
- Extract 88 first-order and gray-level texture features using PyRadiomics
- Include intensity-based statistics, texture features (GLCM, GLRLM, GLSZM), and shape features
- Apply wavelet transforms to capture multi-scale texture patterns
- Model Development:
- Assemble dataset with pre-diagnostic CTs from confirmed PDA cases and matched controls
- Apply least absolute shrinkage and selection operator (LASSO) for feature selection
- Train ensemble classifier (Random Forest or XGBoost) using selected features
- Optimize model using five-fold cross-validation with stratified sampling
- Validation and Interpretation:
- Evaluate model performance on independent test set using AUC, sensitivity, specificity
- Compare AI performance against radiologist interpretation in blinded review
- Generate saliency maps to visualize regions contributing to classification decisions
Quality Assurance: Ensure dataset diversity across scanners and institutions to enhance generalizability. Implement data augmentation techniques to address class imbalance. Adhere to IBSI radiomics standardization guidelines.
Protocol 3: Mass Spectrometry-Based Biomarker Verification
Objective: To verify candidate PDA protein biomarkers using targeted mass spectrometry approaches.
Materials and Reagents:
- High-resolution LC-MS/MS system (Orbitrap or Q-TOF platform)
- C18 reverse-phase chromatography columns
- Trypsin/Lys-C for protein digestion
- Stable isotope-labeled standard peptides
- Solid-phase extraction plates for sample cleanup
- Mobile phase solvents (HPLC-grade water, acetonitrile, methanol)
Methodology:
- Sample Preparation:
- Deplete high-abundance proteins (albumin, IgG) using immunoaffinity columns
- Reduce proteins with dithiothreitol, alkylate with iodoacetamide
- Digest with trypsin (1:20 enzyme-to-protein ratio) overnight at 37°C
- Desalt peptides using C18 solid-phase extraction
- LC-MS/MS Analysis:
- Separate peptides using nanoflow LC with 60-90min gradient (5-35% acetonitrile)
- Operate mass spectrometer in data-dependent acquisition mode for discovery phase
- For targeted verification, implement Parallel Reaction Monitoring (PRM)
- Include stable isotope-labeled internal standards for absolute quantification
- Data Analysis:
- Process discovery data using MaxQuant, PEAKS, or similar platforms
- Identify significantly differentially expressed proteins (p<0.05, fold change >1.5)
- For PRM data, quantify using Skyline software with manual peak curation
- Normalize to internal standards and control samples
- Statistical Validation:
- Assess analytical performance: linearity, limit of detection, precision
- Evaluate clinical performance using ROC analysis on independent sample set
- Compare with CA19-9 and established biomarkers
Quality Control: Include process blanks, pooled quality control samples, and reference standards in each batch. Monitor retention time stability and peak intensity variability (<20% CV).
The integration of advanced methodologies including machine learning-enhanced biomarker panels, artificial intelligence-augmented imaging, and mass spectrometry-based proteomic profiling represents a transformative approach to mitigating false positive and negative results in PDA diagnosis. The documented superiority of multi-marker strategies over single-biomarker reliance, with demonstrated improvements in AUROC from 0.868 to 0.976 for early-stage detection, provides a compelling roadmap for evolving beyond current diagnostic limitations [35]. Similarly, AI-driven radiomics models capable of identifying pre-diagnostic pancreatic changes months before clinical presentation offer unprecedented opportunities for early intervention when treatment efficacy is highest [36]. These approaches collectively address both dimensions of diagnostic inaccuracy—enhancing sensitivity to reduce false negatives while maintaining high specificity to minimize false positives.
Future developments will likely focus on integrating multi-omic data streams to create comprehensive diagnostic models that transcend the limitations of individual methodologies. The combination of circulating biomarker profiles with AI-extracted imaging features and genetic risk markers promises to establish new paradigms for PDA detection, risk stratification, and therapeutic monitoring. Additionally, the translation of mass spectrometry-discovered biomarkers into clinically practical immunoassays will be essential for widespread implementation beyond specialized centers. As these technologies mature, their incorporation into prospective screening trials for high-risk populations will be critical for validating real-world performance and establishing evidence-based guidelines for early detection protocols. Through continued refinement and integration of these advanced approaches, the field moves closer to achieving the fundamental goal of reliable PDA diagnosis at stages when curative interventions remain possible.
Optimizing Spectral Acquisition Parameters and Wavelength Ranges
In the pharmaceutical industry, ensuring the specificity of an analytical method—its ability to measure the analyte accurately in the presence of potential interferents—is a cornerstone of drug quality control and safety assessment. As mandated by the International Council for Harmonisation (ICH), specificity must be established to demonstrate that a method can unequivocally identify and quantify the analyte of interest [1]. This is particularly critical for detecting and characterizing impurities and degradation products, whose presence can compromise patient safety [39].
The combination of Photodiode Array (PDA) detection and Mass Spectrometry (MS) provides an orthogonal, multi-dimensional approach to specificity testing. While PDA detectors can collect full spectra across a range of wavelengths to assess peak purity, MS detection provides unequivocal confirmation based on mass and fragmentation patterns [1]. Modern guidelines, including those from the USP, recommend using peak-purity tests based on PDA or MS to demonstrate specificity conclusively [1]. This application note details optimized spectral acquisition parameters and wavelength ranges within a holistic framework for specificity testing, providing researchers with validated protocols for robust analytical methods.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table catalogues key reagents and materials essential for developing and validating specificity tests using PDA and MS detection.
Table 1: Key Research Reagent Solutions for Specificity Testing
| Reagent/Material | Function & Importance in Specificity Testing |
|---|---|
| Reference Standard | High-purity analyte used for method calibration and as a spectral reference for peak purity assessment [1]. |
| Forced Degradation Reagents | Acids, bases, and oxidizing agents (e.g., HCl, NaOH, H₂O₂) used in stress studies to generate degradation products and challenge method specificity [15]. |
| LC-MS Grade Solvents | High-purity solvents (e.g., Acetonitrile, Water) that minimize background noise and ion suppression in MS, ensuring sensitive and accurate detection [15]. |
| Volatile Buffers | Mobile phase additives (e.g., Ammonium Acetate, Formic Acid) compatible with MS detection that facilitate efficient ion generation and nebulization [15] [39]. |
| Chromatographic Columns | Stationary phases (e.g., C8, C18, F5) that provide the necessary separation to resolve the analyte from its impurities and degradation products [15] [39]. |
Optimized Spectral Acquisition Parameters
Core Principles for Parameter Selection
Optimizing spectral acquisition parameters is fundamental to maximizing the information content of chromatographic data. The core principle is to configure settings that provide high-fidelity data for both quantitative analysis and confident peak identification without introducing artifacts or unnecessary data file sizes. Key considerations include balancing signal-to-noise ratio (S/N) with acquisition speed and ensuring the spectral sampling rate is sufficient to define chromatographic peaks accurately [1]. The following parameters form the foundation of an optimized acquisition method.
Critical Parameters for PDA and MS Detection
Photodiode Array (PDA) Detector Parameters:
- Wavelength Range: Data should be collected across a broad range, typically 190–380 nm or wider, to capture the full UV-Vis profile of the analyte and any potential impurities. This allows for the selection of the optimal wavelength for quantification and provides rich spectral data for peak purity assessment [15].
- Spectral Acquisition Rate: A data sampling rate of 1.56 Hz or higher is recommended to ensure a sufficient number of data points across a chromatographic peak for accurate purity assessment [15].
- Bandwidth and Slit Width: These settings control spectral resolution. Standard settings (e.g., 1 nm) provide a good balance of resolution and S/N for most pharmaceutical applications.
Mass Spectrometry (MS) Parameters:
- Ionization Mode: Electrospray Ionization (ESI) is most common for pharmaceutical compounds. The mode (positive or negative) is selected based on the analyte's chemistry [15].
- Mass Range: A broad mass range (e.g., m/z 100–800) is typically scanned to capture the molecular ions of the analyte, its impurities, and fragments [15].
- Dwell Time: The time spent monitoring a specific ion transition should be optimized (e.g., 100 ms) to ensure sufficient data points per peak while maintaining sensitivity in multi-analyte methods [15].
Table 2: Optimized Spectral Acquisition Parameters for Specificity Testing
| Parameter | PDA Detection | Mass Spectrometry |
|---|---|---|
| Primary Function | Peak Purity, Spectral Identity | Unambiguous Identification, Structural Elucidation |
| Spectral Range | 190–380 nm [15] | m/z 100–800 [15] |
| Data Sampling Rate | ≥ 1.56 Hz [15] | Dwell Time: ~100 ms (MS/MS) [15] |
| Resolution | ~1.2 nm (standard) | High-Resolution (HRAM) or Unit Mass |
| Key Metric | Peak Purity Match Angle/Threshold | Signal-to-Noise (S/N), Mass Accuracy |
Experimental Protocols for Specificity Testing
Comprehensive Specificity and Peak Purity Workflow
A systematic workflow is essential for a thorough specificity study. The following diagram illustrates the integrated protocol using PDA and MS.
Protocol 1: Forced Degradation Studies
Forced degradation studies are performed to validate the stability-indicating nature of the method and to identify likely degradation products [15] [39].
- Sample Preparation: Subject the drug substance and product to stress conditions including:
- Analysis: Analyze stressed samples alongside untreated controls using the optimized LC-PDA-MS method.
- Data Interpretation: Monitor for the appearance of new chromatographic peaks. Use PDA spectra to check for peak homogeneity and MS data to determine the molecular weights and propose structures for degradation products.
Protocol 2: Peak Purity Assessment with PDA Detection
Peak purity assessment is a critical, real-time test for specificity during chromatographic analysis [1].
- Data Collection: Across the entire chromatographic run, the PDA detector collects spectra at multiple points (up-slope, apex, down-slope) for every peak.
- Spectral Comparison: The software algorithmically compares the spectrum at each point in the peak against the spectrum at the peak apex or against a reference standard spectrum.
- Purity Determination: A peak is considered pure if all collected spectra across the peak are identical, within a pre-defined match threshold. A significant spectral difference indicates a co-eluting impurity.
Protocol 3: Orthogonal Identification with Mass Spectrometry
MS provides definitive evidence for the presence of co-eluting compounds that may have different masses but similar PDA spectra [1].
- High-Resolution Mass Spectrometry (HRMS): Use HRMS to obtain exact mass data for the analyte and any impurities. A difference in exact mass, even by a few milliDaltons, confirms the presence of a distinct species.
- Tandem Mass Spectrometry (MS/MS): Fragment the precursor ion. Different compounds will typically produce unique fragmentation patterns, providing structural information and confirming specificity.
Data Analysis and Regulatory Considerations
Validation of the Specificity Method
Once the spectral parameters are optimized and the protocols are executed, the method's performance characteristics must be formally validated per ICH Q2(R2) guidelines [1]. For specificity, this involves demonstrating that the assay is unaffected by the presence of spiked impurities or excipients, and that the analyte peak is free from co-elution [1]. Key parameters to validate include:
- Accuracy: The method should recover a known amount of analyte spiked into a mixture of potential interferents [1].
- Precision: The method should generate reproducible results under normal variations in conditions, known as intermediate precision [1].
- LOD/LOQ: The method's sensitivity for detecting and quantifying impurities must be established, often using signal-to-noise ratios [1].
Advanced Techniques: Scheduled Data-Independent Acquisition
Emerging data acquisition strategies, such as Scheduled Data-Independent Acquisition (SDIA), are being applied to complex analyses like proteomics and can be adapted for impurity tracking. This method uses pre-defined retention time windows for specific ions, which reduces cycle time and eliminates redundant scans. This leads to improved sensitivity and quantitative precision for low-abundance species, making it a powerful technique for monitoring trace-level impurities or degradants [40].
Addressing Co-elution with Similar UV Spectra or Low UV Response
Co-elution, the chromatographic phenomenon where two or more compounds exit the column simultaneously, presents a significant challenge in liquid chromatography (LC) analysis, particularly during pharmaceutical method development and specificity testing. When the co-eluting compounds possess similar ultraviolet (UV) spectral characteristics or when one component has a very low UV response, traditional photodiode array (PDA) detection faces substantial limitations [23] [41]. This application note details advanced methodologies and complementary techniques to overcome these challenges, ensuring accurate peak purity assessment within the framework of procedure for specificity testing.
The fundamental limitation of PDA-based peak purity assessment lies in its dependence on detectable spectral differences between co-eluting compounds. When spectra are nearly identical, as often occurs with structurally related impurities or compounds sharing the same chromophore, the spectral contrast may be insufficient for reliable detection of co-elution [33] [41]. Similarly, low UV-responsive compounds may remain undetected even when spectrally distinct, as their contribution to the combined signal falls below the detection threshold of the PDA system [41]. These scenarios necessitate orthogonal approaches that do not rely solely on UV spectral characteristics for peak purity assessment.
Technical Limitations of PDA Detection
Fundamental Constraints in Peak Purity Assessment
PDA detectors assess peak purity by comparing UV spectra across different points of a chromatographic peak. The underlying principle assumes that a pure peak will exhibit identical spectra throughout its elution profile, while a co-eluting peak will show spectral variations [33]. Commercial chromatography data systems (CDS) calculate metrics such as purity angle and purity threshold to quantify these spectral differences [33] [41]. A peak is typically considered pure when the purity angle is less than the purity threshold [33].
However, this approach encounters significant limitations in specific scenarios:
Similar UV Spectra: Structurally similar compounds, such as those within the same chemical class (e.g., neutral cannabinoids vs. acidic cannabinoids) or compounds sharing the same chromophoric moiety, often exhibit nearly identical UV spectra [23] [41]. Under these conditions, the spectral contrast may be insufficient for the PDA algorithm to detect co-elution, potentially leading to false negative results [41].
Low UV Response: Compounds with low molar absorptivity or those lacking strong chromophores generate weak UV signals [42]. When such compounds co-elute with the main analyte, their spectral contribution may be masked by noise or by the dominant signal of the primary compound, especially when present at low concentrations (<0.1%) [41].
Uniform Co-elution: When impurities co-elute uniformly throughout the entire chromatographic peak (same ratio at both peak base and apex), the spectral shape remains constant across the peak, evading detection by standard purity algorithms [33].
The table below summarizes common scenarios where PDA-based peak purity assessment faces challenges:
Table 1: Limitations of PDA-Based Peak Purity Assessment
| Scenario | Impact on PDA Assessment | Potential Consequence |
|---|---|---|
| Structurally related impurities | Minimal spectral differences; low contrast angle | False negative (co-elution undetected) |
| Low UV-responsive impurities | Weak signal masked by noise or main component | False negative (co-elution undetected) |
| Uniform co-elution profile | Constant spectral shape across peak | False negative (co-elution undetected) |
| High background absorption | Signal distortion, especially at low wavelengths (<210 nm) | False positive (pure peak appears impure) |
| Low concentration analyses | Increased noise-to-signal ratio | Reduced reliability of purity assessment |
Advanced Methodologies and Protocols
Computational Peak Deconvolution Approaches
Advanced computational algorithms can mathematically resolve co-eluted peaks without complete chromatographic separation, leveraging differences in spectral profiles even when minimal.
Protocol: Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) Deconvolution
Data Collection: Acquire chromatographic data using PDA detection with optimal spectral sampling (e.g., 1-2 spectra/second) across the entire UV-Vis range (190-400 nm recommended) [41].
Spectral Preprocessing:
- Apply baseline correction to remove background contributions [43]
- Normalize spectra to account for concentration differences
- Select wavelength ranges with adequate analyte absorbance and minimal mobile phase interference
Model Application:
- Implement MCR-ALS algorithm (available in commercial software such as Shimadzu's LabSolutions with i-PDeA II) [41]
- Define initial estimates of pure component spectra using evolving factor analysis or orthogonal projection approach
- Apply non-negativity constraints to both concentration profiles and spectra
- Iterate until convergence (typically < 0.1% change in residual standard deviation)
Validation:
- Compare deconvoluted spectra with reference standards when available
- Assess residuals for systematic patterns indicating model inadequacy
- Verify quantitative results against known concentrations of spiked standards
This method is particularly effective for resolving co-elutions where compounds have slightly different spectral profiles, as it utilizes the entire spectral information rather than single wavelength monitoring [43] [41].
Protocol: Functional Principal Component Analysis (FPCA) for Large Datasets
For large chromatographic datasets (common in metabolomics or stability studies), FPCA provides an alternative computational approach:
Data Alignment: Perform retention time alignment across all chromatograms in the dataset to address minor shifts [43].
Peak Detection: Identify regions of interest containing co-eluted peaks across multiple samples.
Functional Representation: Convert discrete chromatographic data points into continuous functions using B-spline basis functions [43].
Component Extraction:
- Apply FPCA to detect sub-peaks with the greatest variability across samples
- Identify principal components representing individual compounds within co-eluted peaks
- Generate concentration estimates for each component based on score values
FPCA excels in preserving biologically relevant differences between experimental variants while performing the deconvolution, making it particularly valuable for comparative studies [43].
Mass Spectrometry as an Orthogonal Technique
Mass spectrometry (MS) provides definitive peak purity assessment independent of UV spectral characteristics, making it ideal for addressing PDA limitations [1] [41].
Protocol: LC-MS Peak Purity Assessment
Instrument Setup:
- Employ a single quadrupole MS detector (e.g., Waters QDa, Agilent MSD) coupled to the LC system
- Use electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) source appropriate for the analyte
- Optimize source parameters (temperature, gas flows) for maximum sensitivity
Data Acquisition:
- Monitor relevant ions in simultaneous SIM (Selected Ion Monitoring) and scan mode
- For confirmatory analysis, use tandem mass spectrometry (MS/MS) to monitor specific transitions [44]
- Maintain chromatographic integrity while ensuring MS compatibility (e.g., use volatile buffers)
Peak Purity Assessment:
- Extract ion chromatograms (EIC/XIC) for target analyte and potential impurities [41]
- Compare mass spectra across the chromatographic peak (up-slope, apex, down-slope)
- Verify consistent mass spectral profiles throughout the peak elution
- Confirm presence of expected adducts and isotope patterns across the peak
Data Interpretation:
- Pure Peak: Consistent mass spectra, precursor ions, product ions, and/or adducts across the entire peak [41]
- Impure Peak: Changing mass spectral profiles, detection of unexpected ions, or varying ion ratios across the peak
Table 2: Comparison of Peak Purity Assessment Techniques
| Parameter | PDA Detection | Computational Deconvolution | Mass Spectrometry |
|---|---|---|---|
| Principle | UV spectral contrast | Mathematical separation of spectral profiles | Mass-to-charge ratio separation |
| Sensitivity | Limited by molar absorptivity | Limited by signal-to-noise ratio | High (fg-pg for some analytes) |
| Specificity | Moderate (depends on spectral differences) | Moderate to High | High to Very High |
| Handles similar UV spectra | Poor | Good | Excellent |
| Detects low-UV compounds | Poor | Moderate | Excellent |
| Structural information | Limited (chromophore only) | None | Comprehensive (fragmentation patterns) |
| Resource requirements | Low | Moderate | High |
| Throughput | High | Moderate | Moderate |
Two-Dimensional Liquid Chromatography (2D-LC)
For particularly challenging separations where co-elution persists despite optimization, comprehensive two-dimensional liquid chromatography (LC×LC) provides enhanced separation power by employing two orthogonal separation mechanisms [41].
Protocol: Implementing 2D-LC for Peak Purity Assessment
System Configuration:
- Configure a 2D-LC system with two separate HPLC pumps and a switching valve interface
- Select orthogonal separation mechanisms (e.g., reversed-phase in first dimension, HILIC or ion-exchange in second dimension)
- Optimize interface parameters (loop volume, modulation time) to preserve first-dimension separation
Method Development:
- Establish first-dimension method with optimal resolution but longer run time
- Develop second-dimension method for rapid separation (typically 0.5-2 minutes)
- Balance analysis time with resolution requirements
Data Analysis:
- Create 2D contour plots visualizing retention time in first dimension vs. second dimension
- Identify spots corresponding to pure compounds and potential co-elutions
- Collect spectral or mass data for each spot to confirm identity
Although resource-intensive, 2D-LC provides the highest probability of resolving challenging co-elutions, particularly for complex mixtures like natural products or degradation samples [41].
Experimental Design and Workflow
Integrated Approach to Specificity Testing
A comprehensive specificity testing procedure should strategically combine multiple techniques to overcome the limitations of individual methods. The following workflow provides a systematic approach to address co-elution challenges:
Essential Research Reagent Solutions
Successful implementation of these protocols requires specific reagents and materials designed to address co-elution challenges:
Table 3: Essential Research Reagents and Materials for Addressing Co-elution
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Stable Isotope-Labeled Internal Standards | Corrects for matrix effects; enables accurate quantification in MS | Use structure with 3+ heavy atoms (²H, ¹³C, ¹⁵N); must co-elute with analyte [45] [44] |
| Matrix-Matched Calibrators | Minimizes matrix differences between standards and samples | Prepare in same biological matrix as unknown samples; use stripped matrix for endogenous analytes [45] |
| Spectral Libraries | Reference for computational deconvolution | Create custom libraries with authenticated standards for target compounds [46] |
| Orthogonal Columns | Enhanced separation for 2D-LC | Select different chemistries (C18, HILIC, ion-exchange) for complementary separations [41] |
| Forced Degradation Samples | Generate relevant impurities for method validation | Expose drug substance to stress conditions (heat, light, pH, oxidation) [41] |
| Volatile Mobile Phase Additives | MS-compatible chromatography | Use ammonium formate/acetate, formic/acetic acid instead of non-volatile salts [44] |
Addressing co-elution challenges with similar UV spectra or low UV response requires moving beyond traditional PDA-based peak purity assessment. While PDA detection remains a valuable initial screening tool, its limitations necessitate complementary approaches including computational deconvolution, mass spectrometry, and two-dimensional separation techniques. The integrated workflow presented in this application note provides a systematic strategy for comprehensive specificity testing within pharmaceutical method development.
When implementing these approaches, researchers should consider the specific analytical challenge, available resources, and regulatory requirements. Computational methods offer powerful mathematical solutions without additional hardware but require validation. Mass spectrometry provides definitive identification but at higher operational costs. Two-dimensional LC delivers maximum separation power but with increased method complexity. By understanding the strengths and limitations of each technique, scientists can select the most appropriate strategy to ensure accurate peak purity assessment and develop robust, stability-indicating methods suitable for regulatory submissions.
Managing Baseline Noise, Mobile Phase Effects, and Integration Artifacts
In the pharmaceutical industry, the development of stability-indicating methods is a critical component of regulatory submissions for small-molecule drug candidates. The core of these methodologies lies in effectively managing three fundamental analytical challenges: baseline noise, mobile phase effects, and integration artifacts. These factors directly impact the reliability of specificity testing, particularly when employing Photodiode Array (PDA) and Mass Spectrometry (MS) detection systems. A well-optimized method must demonstrate robust performance across forced degradation studies and routine analysis, ensuring accurate peak purity assessment and precise quantification. This application note provides detailed protocols and structured data to guide researchers in troubleshooting these critical parameters, thereby enhancing method robustness for drug development.
Understanding and Managing Baseline Noise
Baseline noise in chromatographic systems, particularly with PDA detectors, can compromise data quality, lead to failing system suitability tests for noise specifications, and create challenges in quantifying compounds at lower levels [47]. Effective noise management requires a systematic diagnostic and remediation approach.
Troubleshooting Protocol for PDA Baseline Noise
The following workflow provides a step-by-step approach for diagnosing and resolving baseline noise issues in PDA systems. This procedure is applicable to detectors including ACQUITY UPLC PDA, ACQUITY UPLC PDA eλ, and 2998 Photodiode Array detectors [47].
- Step 1: Inspect and Minimize Refractive Index Effects. This is particularly critical when operating at low UV wavelengths (< 205 nm). Key actions include:
- Step 2: Evaluate and Clean the Flow Cell.
- Step 3: Assess the Lamp and Pump.
- Step 4: Apply Software Filters.
- If instrumental troubleshooting does not fully resolve the issue and sensitivity requirements permit, enable the Median Baseline Filter (MBF) in the instrument software to smooth the baseline [47].
Quantitative Data for Baseline Noise Parameters
Table 1: Key Parameters for Baseline Noise Troubleshooting in PDA Systems
| Component | Action | Key Parameter | Reference |
|---|---|---|---|
| Mobile Phase | Sonication | Remove dissolved gases | [47] |
| Flow Cell | Cleaning/Rebuilding | Eliminate particulate/contaminant buildup | [47] |
| Lamp | Replacement | Typical lifetime: ~2000 hours | [47] |
| Back Pressure Regulator | Inspection | Target: ~250 psi | [47] |
| Software Filter | Enable Median Baseline Filter (MBF) | Software-based noise reduction | [47] |
Optimizing Mobile Phase for PDA and MS Detection
The composition of the mobile phase is a critical factor that influences both chromatographic separation and detection sensitivity. Optimization strategies differ between PDA and MS detection systems due to their fundamental operating principles.
Mobile Phase Additive Selection Protocol
This protocol outlines a systematic method for selecting the optimal mobile phase additives to maximize analyte response, particularly for LC-MS applications.
- Step 1: Prioritize Volatility. For LC-MS applications, select only volatile additives that can be efficiently vaporized in the ESI source. Non-volatile salts will cause ion suppression and source contamination [48].
- Step 2: Screen Additives. Prepare separate mobile phases containing different volatile additives at standard concentrations (e.g., 5 mM ammonium acetate, 5 mM ammonium formate, 0.05% formic acid, 0.05% acetic acid) [48].
- Step 3: Analyze and Compare. Inject the analyte of interest using each mobile phase condition on a suitable column (e.g., Ascentis Express F5 for spice cannabinoids) [48].
- Step 4: Evaluate Performance. Assess chromatograms for both chromatographic resolution and relative analyte response (sensitivity). For MS detection, where analytes are discriminated by unique MS/MS transitions, prioritize conditions that yield the highest analyte response [48].
- Step 5: Validate with Sample Matrix. Once the optimal additive is identified, validate the method by analyzing the analyte spiked into the relevant biological matrix (e.g., plasma) following appropriate sample preparation (e.g., HybridSPE-Phospholipid for protein and phospholipid removal) [48].
Experimental Data on Mobile Phase Effects
Table 2: Impact of Mobile Phase Additives on LC-MS Analysis of Spice Cannabinoids
| Mobile Phase Additive | Relative MS Response | Chromatographic Resolution | Notes | Reference |
|---|---|---|---|---|
| 5 mM Ammonium Formate | Highest | Good | Overall best performance for the tested cannabinoids | [48] |
| 0.05% Formic Acid | High | Good | Suitable for acidic conditions | [48] |
| 5 mM Ammonium Acetate | Lower than formate | Good | - | [48] |
| 0.05% Acetic Acid | Lower than formate | Good | - | [48] |
Table 3: Exemplar Chromatographic Conditions for GPB Analysis using LC-PDA
| Parameter | Specification | Reference |
|---|---|---|
| Column | Ascentis Express F5, 2.7 µm, 100 x 4.6 mm | [15] |
| Mobile Phase | 1 mM Ammonium Acetate Buffer (pH ≈5.30) : Acetonitrile (25:75, v/v) | [15] |
| Flow Rate | 0.5 mL/min | [15] |
| System Backpressure | 67 bar | [15] |
| Detection Wavelength | 200 nm | [15] |
| Injection Volume | 1.0 µL | [15] |
| Column Temperature | 40.0 ± 0.1 °C | [15] |
Managing Integration Artifacts and Peak Purity Assessment
Integration artifacts and impure peaks pose a significant risk to the stability-indicating capability of an analytical method. Peak Purity Assessment (PPA) is a critical tool to mitigate the risk of coeluting impurities, which is a core requirement in specificity testing for forced degradation studies [41].
Protocol for Peak Purity Assessment using PDA
This protocol describes the standard procedure for conducting a PDA-facilitated peak purity assessment, a common technique in the pharmaceutical industry.
- Step 1: Acquire Spectral Data. Ensure the PDA detector is configured to collect spectra across a suitable UV range (e.g., 190–380 nm) throughout the entire chromatographic run with an appropriate data sampling rate [15] [41].
- Step 2: Process the Data. Using the CDS software (e.g., Waters Empower, Shimadzu LabSolutions), process the peak of interest. The software algorithm will typically:
- Perform baseline correction by subtracting interpolated baseline spectra between peak liftoff and touchdown [41].
- Convert the spectra into vectors in n-dimensional space and minimize vector lengths using least-squares regression [41].
- Measure the spectral contrast (angle) between the spectrum at the peak apex and every other spectrum across the peak [41].
- Step 3: Interpret Results. The software calculates a purity angle and a purity threshold (a measure of uncertainty due to noise). A peak is generally considered spectrally pure if the purity angle is less than the purity threshold [41].
- Step 4: Review Spectral Overlay. Manually inspect the overlaid spectra from the peak front, apex, and tail for any visually obvious deviations in spectral shape, which indicate a coeluting impurity.
Strengths, Weaknesses, and Artifact Management
PDA-facilitated PPA is efficient and widely accepted, but it is not infallible. Understanding its limitations is crucial for accurate data interpretation [41].
- Strengths:
- Efficient and robust with minimal additional time or resource cost.
- Well-understood and widely used in the pharmaceutical industry [41].
- Weaknesses and False Results:
- False Negatives (Undetected Coelution): Occur when coeluting impurities have very similar UV spectra, poor UV response, elute very near the peak apex, or are present at very low concentrations (<0.1%) [41].
- False Positives (Inhomogeneity for a Pure Peak): Can be caused by significant baseline shifts in gradient elution, suboptimal data processing settings, interference from background noise, measurements at extreme wavelengths (<210 nm), or signals from excipients [41].
Table 4: Alternative Techniques for Peak Purity Assessment
| Technique | Principle | Advantage | Consideration |
|---|---|---|---|
| Mass Spectrometry (MS) | Compare precursor/product ions across the peak in TIC or EIC [41]. | High selectivity and sensitivity. | Requires MS instrumentation; can be more complex. |
| Spiking with Impurity Markers | Co-inject sample with known impurities and observe for peak distortion or new peaks [41]. | Direct and conclusive if markers are available. | Requires authentic impurity standards. |
| Orthogonal Chromatography | Analyze the sample using a chromatographic method with different selectivity (e.g., different column chemistry) [41]. | Can separate coeluting peaks missed by the primary method. | Requires development of a second, validated method. |
| Two-Dimensional LC (2D-LC) | Automatically transfer a fraction of the peak of interest from the first dimension to a second column with orthogonal separation [41]. | Powerful resolving capability. | Requires specialized instrumentation and method development. |
The Scientist's Toolkit: Key Reagents and Materials
Table 5: Essential Research Reagents and Materials for Specificity Testing
| Item | Function/Application | Exemplar Product/Specification |
|---|---|---|
| Volatile Buffers | Mobile phase additives for LC-MS; provide pH control and ionization without signal suppression. | Ammonium acetate, Ammonium formate (5-10 mM) [15] [48]. |
| High-Purity Acids | Mobile phase modifiers for LC-MS and LC-PDA to improve protonation and peak shape. | Formic acid, Acetic acid (0.05-0.1%) [49] [48]. |
| Core-Shell Particle Columns | Provide high-efficiency separations with lower backpressure compared to fully porous sub-2µm particles. | Ascentis Express F5 (2.7 µm) [15] [48]. |
| HILIC Columns | Orthogonal separation mechanism for polar compounds that are poorly retained in reversed-phase mode. | PC HILIC column (e.g., for NMN analysis) [49]. |
| Phospholipid Removal SPE | Selectively removes phospholipids from biological samples (e.g., plasma) to reduce ion suppression in LC-MS. | HybridSPE-Phospholipid 96-well plates [48]. |
| Strong Basic Anion Exchange Resin | Selective clean-up of complex natural extracts; removes chlorophylls while retaining carotenoids. | Ambersep 900 OH resin [50]. |
Concentration Optimization for Reliable Peak Purity Assessment
Within the framework of specificity testing for pharmaceutical analysis, reliable peak purity assessment is a critical component. It ensures that the primary analyte chromatographic peak is free from coeluting impurities, which is essential for accurate quantification and method validation [51]. This application note details a systematic, evidence-based protocol for optimizing sample concentration to achieve dependable peak purity assessments using Photodiode Array (PDA) and Mass Spectrometry (MS) detectors. The fundamental principle is that an improperly chosen concentration—either too high or too low—can compromise the purity assessment, leading to false positives or false negatives [51] [52]. We provide a detailed, executable methodology for establishing the optimal concentration window, thereby enhancing the reliability of specificity testing procedures.
Principles of Peak Purity Assessment
Peak purity assessment is a qualitative tool that evaluates the spectral homogeneity of a chromatographic peak. The underlying principle is that a pure compound will exhibit identical UV-Vis spectra across all points of the peak (at the peak apex, and on the leading and trailing edges), whereas a coeluting impurity will cause spectral variations [51].
- PDA-Based Assessment: A PDA detector is the most common tool for this purpose. It collects full spectra continuously across the peak. The software then calculates metrics such as the purity angle and purity threshold. A purity angle less than the purity threshold suggests a spectrally pure peak. However, this should never be the sole criterion for final judgment [51].
- MS-Based Assessment: Liquid Chromatography-Mass Spectrometry (LC-MS) provides a more definitive, orthogonal assessment by detecting coelution based on mass differences. It is particularly powerful for identifying low-level contaminants that may not have a distinct UV spectrum [51] [53].
A significant limitation of UV-based assessment is its dependence on detector sensitivity and the spectral contrast between the analyte and the impurity. If the impurity has a very similar UV spectrum or is present at a very low level, the PDA detector may not detect it, resulting in a false pure assessment [51]. The concentration of the analyte directly influences the signal-to-noise ratio and the detector's ability to identify these subtle spectral differences, making its optimization paramount.
Critical Parameters for Concentration Optimization
The goal of concentration optimization is to find the range where the analyte response is linear and the detector can accurately identify spectral inhomogeneity without being overwhelmed by noise or signal saturation. The following parameters are crucial for this process.
Detector Parameter Optimization
Before finalizing sample concentration, key detector parameters must be optimized to ensure the system is capable of high-fidelity data acquisition. These parameters are interdependent and can significantly impact the perceived purity [52].
Table 1: Key HPLC/PDA Detector Parameters for Optimization
| Parameter | Function | Optimization Consideration | Impact on Peak Purity |
|---|---|---|---|
| Data Rate | Speed of data collection (Hz) | Set to collect 25-50 points across the narrowest peak. Insufficient points poorly define peaks; excessive rates increase noise [52]. | Affects peak definition and accuracy of spectral collection across the peak. |
| Filter Time Constant | Electronic noise filter | Slower settings reduce baseline noise but can broaden peaks. A balanced setting is required to minimize noise without compromising peak shape [52]. | Reduces high-frequency noise, improving spectral clarity and purity algorithm performance. |
| Slit Width | Controls light reaching the PDA | Wider slits increase light throughput and sensitivity but decrease spectral resolution. Narrower slits improve resolution at the cost of signal [52]. | Influences spectral resolution; critical for distinguishing impurities with similar UV spectra. |
| Spectral Resolution (Bandwidth) | Number of diodes averaged per data point | Higher values (e.g., 4-8 nm) reduce noise but decrease spectral detail. Lower values (e.g., 1-2 nm) provide high spectral fidelity [52]. | Determines the level of detail in each collected spectrum, which is vital for detecting subtle spectral shifts. |
| Absorbance Compensation | Reduces non-wavelength specific noise | Uses a baseline wavelength region to subtract background noise, improving the signal-to-noise ratio [52]. | Enhances the signal-to-noise ratio, allowing for more sensitive detection of minor spectral contributions. |
Defining the Analytical Target Profile (ATP) and Critical Quality Attributes (CQAs)
A systematic approach, such as Analytical Quality by Design (AQbD), should be employed. The process begins by defining the Analytical Target Profile (ATP), which states the overall objective of the method [54]. For peak purity assessment, the ATP can be defined as: "The method must reliably detect a specified impurity (e.g., 0.1%) coeluting with the main peak."
From the ATP, the Critical Quality Attributes (CQAs) are identified. These are the measurable metrics that define method performance [54]. For this application, the key CQAs are:
- Peak Purity Match Score: A numerical value (e.g., >990) indicating spectral homogeneity.
- Signal-to-Noise Ratio (S/N): A minimum S/N (e.g., ≥10) for the analyte peak is required for reliable spectral comparison [52].
- Spectral Contrast: The ability of the method to distinguish the analyte spectrum from potential impurities.
Experimental Protocol: Concentration Optimization Workflow
This protocol provides a step-by-step guide for determining the optimal concentration range for peak purity assessment.
Materials and Reagents
Table 2: Research Reagent Solutions and Essential Materials
| Item | Specification / Function |
|---|---|
| HPLC System | Alliance iS HPLC System or equivalent, equipped with a PDA detector and auto-sampler [52]. |
| Analytical Column | XBridge BEH C18, 250 x 4.6 mm, 5 µm or equivalent reversed-phase column [52]. |
| MS System (Orthogonal) | LC-MS/MS system (e.g., QDa Mass Detector) for confirmatory analysis [53] [54]. |
| Reference Standard | High-purity analyte reference standard. |
| Impurity Standards | Chemically related compounds or known degradation products. |
| Mobile Phase | HPLC-grade solvents and buffers, prepared as per method requirements (e.g., 4g/L Chloroacetic Acid in 40:60 water:acetonitrile, pH 3.0) [52]. |
| Diluent | A solvent that fully dissolves the analyte and is compatible with the mobile phase. |
Step-by-Step Procedure
Preparation of Stock and Working Solutions
- Prepare a stock solution of the analyte at a high concentration (e.g., 1 mg/mL).
- Perform a series of serial dilutions to create working solutions covering a broad concentration range (e.g., 0.1 µg/mL, 1 µg/mL, 10 µg/mL, 50 µg/mL, 100 µg/mL, 200 µg/mL).
Instrumental Setup and Parameter Configuration
- Install and condition the chromatographic column as per manufacturer's instructions.
- Set the mobile phase composition, flow rate, and column temperature according to the separation method.
- Configure the PDA detector. Initially, use a medium data rate (e.g., 2-10 Hz), a normal filter time constant, a slit width of 50 µm, and a resolution of 4 nm as a starting point [52]. Set the wavelength for monitoring and the spectral collection range (e.g., 210-400 nm).
Chromatographic Analysis
- Inject each concentration in the series in triplicate.
- For orthogonal confirmation, analyze key concentrations using the LC-MS system with electrospray ionization in both positive and negative modes [53].
Data Analysis and Determination of Optimal Concentration
- For each chromatographic run, measure the signal-to-noise ratio (S/N) for the main peak.
- Use the PDA software to calculate the peak purity match score or purity angle/threshold.
- For the LC-MS data, examine the extracted ion chromatograms for the main analyte and potential impurities for any signs of coelution.
Workflow Visualization
The following diagram illustrates the logical workflow for the concentration optimization experiment.
Data Interpretation and Acceptance Criteria
The data collected from the concentration series must be systematically evaluated against pre-defined acceptance criteria.
Table 3: Quantitative Data from a Hypothetical Concentration Optimization Study
| Concentration (µg/mL) | Avg. S/N Ratio | Avg. Peak Purity Match (0-1000) | PDA Assessment | MS Assessment | Conclusion |
|---|---|---|---|---|---|
| 0.1 | 5 | 850 | Unreliable (Low S/N) | Not Detected | Too Low |
| 1.0 | 50 | 995 | Pure | No Impurity Detected | Lower Limit |
| 10.0 | 500 | 998 | Pure | No Impurity Detected | Optimal |
| 50.0 | 2500 | 995 | Pure | No Impurity Detected | Optimal |
| 100.0 | 5000 | 950 | Impure (Spectral Shift) | Impurity Ion Detected | Saturation |
| 200.0 | 9000 | 800 | Impure (Spectral Shift) | Impurity Ion Detected | Too High |
Interpretation of Results:
- Low Concentration Range (0.1 - 1.0 µg/mL): The S/N ratio is below the acceptable threshold (e.g., NLT 10), making the spectral data too noisy for a reliable purity assessment [52]. The lower limit of the optimal range is the concentration where the S/N consistently exceeds the threshold and the purity score stabilizes at a high value.
- Optimal Concentration Range (10.0 - 50.0 µg/mL): The S/N ratio is strong, and the peak purity match score is high and consistent. The PDA spectra are homogeneous, and the orthogonal MS data confirms the absence of coeluting impurities. This is the target range for routine peak purity assessment.
- High Concentration Range (100.0 - 200.0 µg/mL): The decreasing purity match score indicates a loss of spectral homogeneity. This is often due to detector saturation or overloading of the chromatographic column, which can cause peak distortion and reveal minor spectral contributions from impurities or solvent effects. The analyte peak is correctly flagged as "impure" despite the high S/N.
Integrated Peak Purity Assessment Protocol
Once the optimal concentration is established, the following integrated protocol should be used for reliable peak purity assessment in specificity testing.
Procedure:
- Sample Analysis: Analyze the test sample at the pre-determined optimal concentration using the optimized HPLC-PDA method.
- Software-Assisted Purity Assessment: For the peak of interest, run the peak purity algorithm in the CDS software to obtain the purity angle and threshold.
- Critical Manual Review: Regardless of the software result, manually review the spectral overlays from the peak apex, leading edge, and trailing edge. Look for any subtle shifts or shoulders in the spectra that the algorithm may have missed [51]. This step is non-negotiable.
- Orthogonal Confirmation: If any ambiguity arises from the PDA assessment, or as a required part of method validation, perform analysis by LC-MS. The mass-based detection will provide definitive evidence of coelution [51] [53] [54].
- Final Judgment: A peak is confirmed pure only when both PDA assessment (after manual review) and MS analysis (if performed) confirm the absence of coeluting substances.
This application note establishes that concentration is a foundational parameter for reliable peak purity assessment. A systematic optimization process, moving beyond default instrument settings and exploring a wide concentration range, is essential. By defining an optimal concentration window that avoids the pitfalls of low S/N and detector saturation, and by integrating PDA with manual spectral review and orthogonal MS confirmation, analysts can achieve a true and reliable assessment of peak purity. This robust approach significantly strengthens specificity testing protocols within drug development, ensuring data integrity and regulatory compliance.
Validation Strategies and Orthogonal Technique Integration
Establishing Acceptance Criteria for Specificity Demonstration
Specificity is a fundamental parameter in analytical method validation, confirming that a method can accurately measure the analyte of interest in the presence of other components that may be expected to be present in the sample, such as impurities, degradants, or excipients [1]. In the context of modern pharmaceutical analysis, specificity is typically demonstrated using techniques like photodiode array (PDA) detection and mass spectrometry (MS), which provide orthogonal means of assessing peak purity and detecting potential interferences [41]. This document establishes comprehensive acceptance criteria and detailed protocols for demonstrating specificity, framed within the broader procedure for specificity testing using PDA and mass spectrometry research.
Regulatory and Scientific Framework
Regulatory guidelines, including those from the International Council for Harmonisation (ICH), require that analytical procedures include investigations to demonstrate specificity [1]. While ICH Q2(R1) notes that "peak purity tests may be useful to show that the analyte chromatographic peak is not attributable to more than one component," it does not mandate a single technique for this demonstration [41]. The primary goal is to establish, through laboratory studies, that the performance characteristics of the method meet the requirements for the intended analytical application, providing assurance of reliability during normal use [1]. It is critical to understand that peak purity assessment never unequivocally proves a peak is pure; rather, it can only conclude that no co-eluted compounds were detected under the testing conditions [41].
Acceptance Criteria for Specificity Demonstration
General Specificity Requirements
For a method to be considered specific, it must demonstrate the ability to measure the analyte accurately and specifically in the presence of all potential sample components. Key acceptance criteria include:
- Chromatographic Separation: Resolution factor (Rs) ≥ 2.0 between the analyte and the closest eluting potential interferent [1]
- System Suitability: Tailing factor ≤ 2.0 and theoretical plate count ≥ 2000 for the analyte peak [1]
- Forced Degradation Studies: Mass balance of 98.0-102.0% for stressed samples to account for all degradation products [1]
- Interference Check: No interference observed from blank, placebo, or known impurities at the retention time of the analyte [1]
PDA-Based Peak Purity Acceptance Criteria
PDA-facilitated peak purity assessment examines changes in the UV absorbance spectrum throughout the peak to detect co-eluted compounds with different UV absorbance spectra [41].
Table 1: Acceptance Criteria for PDA-Based Peak Purity Assessment
| Parameter | Acceptance Criterion | Calculation Method | Remarks |
|---|---|---|---|
| Purity Angle | Must be less than the Purity Threshold | Spectral contrast angle between all spectra within a peak and the apex spectrum [41] | Applied to stressed samples and known impurities |
| Spectral Similarity | Match factor ≥ 990 (or 0.990) when comparing spectra across the peak | Normalized spectral comparison using cosine or correlation algorithms [41] | Values may vary between software platforms |
| Purity Threshold | Algorithmically determined value accounting for solvent and noise contributions | Based on variation in spectral vector and system noise [41] | Provides the uncertainty margin for purity assessment |
Mass Spectrometry-Based Specificity Criteria
MS detection provides complementary specificity assessment through mass-to-charge ratio monitoring rather than spectral characteristics.
Table 2: Acceptance Criteria for Mass Spectrometry-Based Specificity Assessment
| Parameter | Acceptance Criterion | Application | Considerations |
|---|---|---|---|
| Extracted Ion Chromatogram (XIC) Purity | Consistent mass spectral profile across the entire chromatographic peak [41] | Nominal mass resolution instruments (e.g., single quadrupole) | Precursor ions, product ions, and adducts should remain constant |
| Mass Accuracy | ≤ 5 ppm for high-resolution MS | HRMS systems (Q-TOF, Orbitrap) | Confirms elemental composition |
| Ion Ratio Consistency | ± 20-30% relative for product ion ratios across the peak | MS/MS systems (QqQ, Q-TOF) | Verifies consistent fragmentation pattern |
Experimental Protocols
Comprehensive Specificity Demonstration Workflow
The following diagram illustrates the complete workflow for establishing method specificity using orthogonal techniques:
Forced Degradation Study Protocol
Forced degradation studies provide critical evidence of method specificity by demonstrating separation of degradation products from the main analyte.
Materials and Reagents:
- Drug substance/drug product
- Appropriate solvents (water, organic solvents as required)
- Acid (e.g., 0.1N HCl)
- Base (e.g., 0.1N NaOH)
- Oxidizing agent (e.g., 3% H₂O₂)
- Thermal chamber
- Photostability chamber
Procedure:
- Sample Preparation: Prepare separate samples of drug substance or drug product at target concentration
- Stress Conditions Application:
- Acidic Hydrolysis: Expose to 0.1N HCl at 60°C for 1-7 days
- Basic Hydrolysis: Expose to 0.1N NaOH at 60°C for 1-7 days
- Oxidative Degradation: Treat with 3% H₂O₂ at room temperature for 24 hours
- Thermal Degradation: Heat solid sample at 80°C for 1-2 weeks
- Photolytic Degradation: Expose to appropriate light conditions (e.g., 1.2 million lux hours)
- Sample Analysis: Analyze stressed samples alongside appropriate controls
- Assessment:
- Determine extent of degradation (typically 5-20% degradation is targeted)
- Evaluate mass balance
- Perform peak purity assessment on main peak
- Identify and characterize degradation products
PDA Peak Purity Assessment Protocol
Instrumentation: HPLC system with photodiode array detector
Procedure:
- Method Setup: Ensure appropriate spectral acquisition parameters:
- Wavelength range: 210-400 nm (or appropriate range for analyte)
- Spectral acquisition rate: ≥ 10 spectra per peak
- Resolution: 1-2 nm
- Data Acquisition: Inject and analyze samples, ensuring proper peak shape and adequate signal-to-noise ratio
- Peak Purity Analysis:
- Select analyte peak for purity assessment
- Define peak start and end points using valley-to-valley integration
- Process spectra with appropriate baseline correction
- Review purity plot and calculated purity parameters
- Interpretation: Compare purity angle to purity threshold; peak is considered pure if purity angle < purity threshold
Mass Spectrometry Peak Purity Protocol
Instrumentation: LC-MS system with appropriate ionization source and mass analyzer
Procedure:
- Method Setup:
- Ionization mode: ESI or APCI (positive/negative as appropriate for analyte)
- Mass range: Appropriate for analyte and potential impurities
- Scan speed: Adequate to capture multiple scans across chromatographic peak
- Data Acquisition:
- For full scan MS: Acquire data across appropriate m/z range
- For MS/MS: Monitor specific precursor → product ion transitions
- Peak Purity Assessment:
- Extract ion chromatograms for analyte and potential impurities
- Compare mass spectra across the chromatographic peak (at peak front, apex, and tail)
- Assess consistency of mass spectral profile
- Interpretation: Consistent mass spectral profile across the peak indicates peak purity
Research Reagent Solutions and Essential Materials
Table 3: Essential Materials for Specificity Demonstration Studies
| Category | Item | Function/Application | Specifications/Considerations |
|---|---|---|---|
| Chromatography | HPLC/UHPLC System | Separation of analytes from potential interferents | Compatibility with detection systems (PDA, MS) |
| Analytical Column | Chromatographic separation | Appropriate chemistry (C18, phenyl, etc.), particle size, dimensions | |
| Mobile Phase Components | Solvent system for separation | HPLC-grade solvents, high-purity buffers and additives | |
| Detection | Photodiode Array Detector | UV spectral acquisition for peak purity assessment | Wavelength range, spectral resolution, acquisition rate [41] |
| Mass Spectrometer | Mass-based detection and peak purity assessment | Appropriate mass analyzer (single quadrupole, Q-TOF, etc.), ionization source [4] | |
| Sample Preparation | Reference Standards | Method development and qualification | Certified purity, appropriate documentation |
| Chemical Stress Reagents | Forced degradation studies | Acids, bases, oxidizing agents of appropriate concentration and purity [1] | |
| Software | Chromatography Data System (CDS) | Data acquisition and processing | Peak purity algorithm capabilities (e.g., Empower, OpenLab, LabSolutions) [41] |
| Mass Spectrometry Software | MS data acquisition and interpretation | Qualitative and quantitative analysis capabilities |
Orthogonal Assessment and Advanced Techniques
Two-Dimensional Liquid Chromatography (2D-LC)
For complex specificity challenges, two-dimensional liquid chromatography provides enhanced separation power by combining two independent separation mechanisms.
Application: Particularly valuable for complex matrices where single-dimension separation may be insufficient to resolve all components.
Peak Purity Assessment Limitations and Mitigation Strategies
Both PDA- and MS-based peak purity assessments have limitations that must be considered when interpreting results:
Table 4: Limitations of Peak Purity Assessment Techniques and Mitigation Strategies
| Technique | Limitations | False Negative/Positive Risks | Mitigation Strategies |
|---|---|---|---|
| PDA-Based PPA | - Co-eluting impurities with minimal spectral differences [41] - Poor UV response of impurities [41] - Impurities eluting near peak apex [41] | False Negative: PPA indicates purity despite co-elution [41] False Positive: PPA indicates impurity with pure peak [41] | - Optimize spectral acquisition parameters - Use complementary techniques (MS) - Employ orthogonal chromatography |
| MS-Based PPA | - Similar fragmentation patterns - Ion suppression effects - Concentration-dependent detection | False Negative: Co-eluting compounds with similar mass spectra False Positive: In-source fragmentation or adduct formation | - Vary ionization parameters - Use high-resolution MS - Employ different ionization modes |
Documentation and Regulatory Considerations
Comprehensive documentation of specificity demonstration is essential for regulatory submissions. The following elements should be included:
- Protocol: Pre-approved study protocol with predefined acceptance criteria
- Raw Data: Chromatrograms, spectra, and peak purity plots for all determinations
- Summary Reports: Tabulated results with clear pass/fail assessment against acceptance criteria
- Justification: Scientific rationale for selected conditions and any deviations from target conditions
- Conclusion: Clear statement on method specificity with supporting evidence
Specificity demonstration should be an iterative process during method development, with refinement based on findings from forced degradation studies and peak purity assessments. The combination of PDA and mass spectrometry provides complementary techniques that collectively build a compelling case for method specificity, ultimately ensuring reliable measurement of the analyte in the presence of potential interferents throughout the method's lifecycle.
In the field of analytical chemistry, ensuring the specificity of a method is paramount, particularly in drug development and bioanalysis. High-Performance Liquid Chromatography (HPLC) and Ultra-Performance Liquid Chromatography (UPLC) are separation mainstays, but the choice of detector defines the depth and quality of the information obtained. Two detectors with distinct advantages are the Photodiode Array (PDA) detector and the Mass Spectrometric (MS) detector. The PDA detector provides ultraviolet-visible (UV-Vis) spectral data, while the MS detector provides mass-to-charge ratio information. This application note delineates their complementary strengths, providing a structured comparison and detailed protocols to guide researchers in deploying these techniques for robust specificity testing. Data presented herein is framed within a broader thesis on analytical procedure development, underscoring how the synergistic use of PDA and MS detection enhances method reliability and information richness for critical applications in pharmaceutical and nutritional sciences.
Comparative Detector Performance: Quantitative Data
The fundamental differences between PDA and MS detectors lead to significant variations in their analytical performance, particularly concerning sensitivity and susceptibility to matrix effects. A direct comparison for the analysis of lipophilic micronutrients and carotenoids in biological samples illustrates this point.
Table 1: Comparative Sensitivity of HPLC-PDA and HPLC-MS/MS for Selected Analytes [55]
| Analyte | Detection Method | Relative Sensitivity | Notes |
|---|---|---|---|
| Lycopene | HPLC-MS/MS | Up to 37x more sensitive than PDA | - |
| α-Carotene | HPLC-MS/MS | Up to 37x more sensitive than PDA | Exhibited matrix suppression in MS/MS |
| β-Carotene | HPLC-MS/MS | Up to 37x more sensitive than PDA | Exhibited matrix suppression in MS/MS |
| Lutein | HPLC-PDA | Up to 8x more sensitive than MS/MS | MS/MS signal enhanced by matrix |
| β-Cryptoxanthin | HPLC-MS/MS | Sensitivity data not specified | MS/MS signal enhanced by matrix |
| α-Tocopherol | Both | Similar suitability | - |
| Retinyl Palmitate | Both | Similar suitability | Exhibited matrix suppression in MS/MS |
Key Observations from Quantitative Data: [55]
- MS/MS Superior Sensitivity: For the majority of analytes tested (lycopene, α-carotene, and β-carotene), HPLC-MS/MS demonstrated significantly higher sensitivity, being up to 37 times more sensitive than HPLC-PDA. This is crucial for quantifying compounds at low biological concentrations, thereby minimizing necessary blood sample volumes in clinical studies.
- PDA Advantages for Specific Compounds: PDA detection proved more sensitive for certain compounds like lutein, highlighting that the optimal detector is application-dependent.
- Matrix Effects: The study revealed significant matrix effects that differentially impacted the detectors. MS/MS signals were enhanced by matrix components for lutein and β-cryptoxanthin but suppressed for retinyl palmitate, α-carotene, and β-carotene. In contrast, the PDA signal is matrix-independent, providing a more reliable quantification in these specific cases.
- Unique Capabilities of MS/MS: The MS/MS detector exclusively allowed for the quantitation of minor components such as phylloquinone, (Z)-lycopene isomers, and several minor retinyl esters, which were not feasible to quantify using PDA detection alone [55].
Experimental Protocols
The following protocols are adapted from validated methods used for the analysis of fat-soluble vitamins and carotenoids in human plasma chylomicron fractions and for the characterization of pigments in microalgae.
Protocol 1: HPLC-PDA and HPLC-MS/MS for Fat-Soluble Micronutrients
This protocol outlines the simultaneous extraction and analysis of carotenoids, tocopherols, and retinyl esters from triglyceride-rich lipoprotein (TRL) fractions, enabling a direct comparison of PDA and MS/MS detection [55].
I. Sample Preparation (Chylomicron Isolation & Extraction)
- Plasma Separation: Draw venous blood into EDTA vacutainer tubes. Centrifuge at 1000-1700 g at 4°C for 10 minutes to separate plasma from red blood cells. Perform all subsequent steps under subdued light.
- Ultracentrifugation: Transfer 2.5 mL of plasma to a PVA-coated polyallomer tube. Overlayer with 1 mL of NaCl solution (density 1.006 kg/L). Subject to ultracentrifugation at 40,700 rpm for 33 minutes in a swinging bucket rotor.
- TRL Collection: Collect the top 0.5 mL supernatant (TRL fraction). Rinse the tube with 250 μL saline and combine with the fraction.
- Lipid Extraction:
- Mix a 0.5 mL aliquot of the TRL fraction with 0.5 mL ethanol.
- Add 2 mL of extraction solvent (e.g., Hexane/Ethanol/Acetone/Toluene, 10:6:7:7, v/v/v/v).
- Probe-sonicate for 8 seconds, repeat twice.
- Centrifuge at 300 g for 5 minutes.
- Transfer the upper non-polar layer.
- Re-extract the aqueous phase once more and combine the non-polar extracts.
- Evaporate the combined extract to dryness under a stream of nitrogen at <25°C.
- Reconstitute the dried extract in an appropriate injection solvent.
II. Instrumental Analysis
Table 2: HPLC-PDA-MS/MS Instrument Parameters [55]
| Parameter | HPLC-PDA Configuration | HPLC-MS/MS Configuration |
|---|---|---|
| HPLC System | Agilent 1200 Series | Agilent 1200 Series |
| Column | C18 reversed-phase | C18 reversed-phase |
| Detection | Photodiode Array (PDA) | QTRAP 5500 Mass Spectrometer |
| Ion Source | Not Applicable | Atmospheric Pressure Chemical Ionisation (APCI) |
| Ion Mode | Not Applicable | Positive |
| Data Acquisition | UV-Vis spectra (e.g., 190-800 nm), multiple wavelengths | Multiple Reaction Monitoring (MRM) |
III. Data Analysis for Specificity
- PDA Specificity Assessment: Overlay UV-Vis spectra of analyte peaks at upslope, apex, and downslope. Peak homogeneity is confirmed by a high spectral match (>99%), indicating a pure peak. Spectral differences suggest a co-eluting impurity [56].
- MS/MS Specificity Assessment: Specificity is confirmed by the unique precursor ion → product ion transition(s) for each analyte and a stable retention time matching the standard. The MRM mode provides inherent specificity by filtering for a specific mass fragment.
Protocol 2: HPLC-PDA-MS/MS for Pigment Profiling in Microalgae
This protocol describes an integrated approach for the identification and quantification of carotenoids and chlorophylls, leveraging the strengths of both detectors in a single run [53].
- Sample Extraction: Extract freeze-dried microalgae biomass with a suitable solvent (e.g., methanol) until the residue becomes colorless.
- HPLC-PDA-MS/MS Analysis:
- Chromatography: Use a reversed-phase C18 column with a gradient elution of water and acetonitrile/methanol.
- Detection in Series: The HPLC effluent first passes through the PDA detector, then into the MS spectrometer.
- PDA Data Collection: Collect full UV-Vis spectra (e.g., 190-800 nm) for all peaks.
- MS Data Collection: Operate the MS with Electrospray Ionisation (ESI) or APCI in positive/negative mode. Use full scans for discovery and MRM for quantification.
IV. Specificity and Identification [53] [57]
- Identification: Assign compound identities by matching retention times, UV-Vis spectral characteristics (from PDA), and mass spectra (from MS/MS) against those of authentic standards.
- Purity Assessment: Use the PDA spectrum to assess peak homogeneity.
- Sensitivity: For quantification of major pigments (lutein, β-carotene), PDA data is often sufficient. For low-abundance or co-eluting compounds, the superior sensitivity and specificity of MS/MS are utilized.
Workflow Visualization
The following diagram synthesizes the logical decision process for employing PDA and MS detection within an analytical procedure for specificity testing.
Analytical Specificity Testing Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details key materials and reagents essential for conducting the experiments described in the protocols above.
Table 3: Essential Research Reagents and Materials [55] [53] [4]
| Item | Function / Application | Example in Protocol |
|---|---|---|
| C18 Reversed-Phase HPLC Column | Chromatographic separation of analytes based on hydrophobicity. | Separation of carotenoids, tocopherols, and retinyl esters [55]. |
| Authentic Analytical Standards | Method development, calibration, and identification by matching retention time and spectral data. | Lutein, β-carotene, α-tocopherol, retinol for quantification [55]. |
| Stable Isotope-Labeled Internal Standards | Correct for analyte loss during extraction and matrix effects in MS/MS. | d8-β-Carotene for MS/MS analysis [55]. |
| HPLC-grade Solvents | Mobile phase preparation and sample extraction to minimize background interference. | MTBE, hexane, ethanol, methanol, acetonitrile [55]. |
| Mass Spectrometry Tuning Solution | Calibration and performance optimization of the mass spectrometer. | Specific to MS instrument manufacturer (e.g., ESI/APCI tuning mix). |
| Lipid Extraction Solvent Mixtures | Efficient extraction of lipophilic compounds from complex biological matrices. | Hexane/Ethanol/Acetone/Toluene for TRL fraction extraction [55]. |
In pharmaceutical analysis, specificity is the ability of a method to measure accurately and specifically the analyte of interest in the presence of other components that may be expected to be present in the sample, such as impurities, degradants, or excipients [1]. Demonstrating specificity is a critical requirement for stability-indicating methods and is mandated by regulatory bodies for method validation. The integration of orthogonal techniques—methods based on different separation or detection mechanisms—provides the highest confidence in demonstrating method specificity [41]. This application note details practical protocols for combining two powerful orthogonal approaches: two-dimensional liquid chromatography (2D-LC) and spiking studies, within the framework of specificity testing using photodiode array (PDA) and mass spectrometry (MS) detection.
The fundamental principle behind using orthogonal methods is that no single analytical technique is infallible. While PDA-based peak purity assessment is the most common approach for demonstrating specificity, it has limitations, including potential false negatives when co-eluted impurities have minimal spectral differences or poor UV responses [41]. Combining PDA with MS detection and orthogonal separation techniques like 2D-LC provides a multi-layered approach that significantly enhances the reliability of specificity assessments for small molecule drug candidates.
Theoretical Foundations of Orthogonality
The Concept of Orthogonality in Separation Science
In comprehensive two-dimensional liquid chromatography (LC×LC), the degree of orthogonality between both dimensions is a critical factor for obtaining higher peak capacities [58]. A 2D LC separation is considered "orthogonal" if the two separation mechanisms are independent of each other, therefore providing complementary selectivities [58]. This orthogonality allows sample components to be spread out through two different retention patterns, significantly enhancing the probability of resolving closely eluting compounds that might co-elute in a single-dimensional separation.
Successful orthogonal separations can be achieved when suitable mobile and stationary phases are selected, taking into account the physicochemical properties of the sample components including size and charge, hydrophobicity and polarity [58]. LC techniques offer a wide variety of separation mechanisms, such as normal-phase (NP), reversed-phase (RP), size-exclusion (SEC), ion exchange (IEX), and affinity chromatography (AC), which are characterized by different selectivities [58]. The combination of HILIC and reversed-phase conditions in the 1D and 2D, respectively, has emerged as a particularly promising orthogonal combination [58].
Detector Orthogonality: PDA and MS
The complementary nature of PDA and MS detectors provides a powerful orthogonal approach for specificity testing. PDA detectors collect full UV spectra across a range of wavelengths throughout the peak, enabling spectral contrast analysis to detect co-eluting compounds with different UV profiles [41]. Mass spectrometry, particularly when used with single quadrupole or tandem mass spectrometers, assesses peak purity by demonstrating the presence of the same precursor ions, product ions, and/or adducts across the peak attributed to the parent compound [41].
Research has demonstrated that combining data from PDA and MS detectors leads to superior classification results compared to using either detection method alone. In studies analyzing genuine and counterfeit medicines, the combination of PDA and MS data resulted in fewer classification errors between genuine/generic and counterfeit products compared to PDA or MS data separately [24]. This detector orthogonality is particularly valuable for mitigating the risk of false negatives in peak purity assessments.
Table 1: Comparison of PDA and MS Detection Capabilities for Specificity Testing
| Parameter | PDA Detection | MS Detection |
|---|---|---|
| Primary Principle | Spectral contrast analysis throughout peak | Mass spectral consistency across peak |
| Key Metrics | Purity angle vs. threshold; Spectral similarity | Ion ratios; Mass chromatographic profiles |
| Strengths | Non-destructive; Well-understood in industry; Minimal extra cost | High sensitivity; Structural information; Handles similar UV spectra |
| Limitations | Limited for compounds with similar UV spectra; Low UV response issues | Solvent incompatibilities; Ion suppression possible |
| False Negative Risk | When co-eluters have similar UV spectra | When co-eluters have similar mass fragmentation |
| Regulatory Status | De facto standard for many applications | Increasingly accepted as orthogonal approach |
Comprehensive Two-Dimensional Liquid Chromatography (2D-LC)
Instrumentation and Method Development
An LC×LC system is composed of at least two pumps, two columns, an injector, an interface, and a detector [58]. The interface hyphenates the two dimensions, typically using a multi-port switching valve with storage loops that alternately collect effluent from the first dimension and transfer it to the second dimension [58]. The most widely used interface involves a 2-position/10-port switching valve, though 2-position/8-port valves or two 2-position/6-port valves have also been implemented [58].
Method development for LC×LC requires careful optimization of several parameters. For the second dimension analyses, separation time should be fast enough to ensure complete fraction elution before subsequent transfer and adequate first dimension sampling [58]. This time is particularly critical and may be enhanced if a regeneration step is needed when running gradient programs. After defining the 2D analysis time, the 1D analysis time can be optimized at low flow rates to achieve three to four samplings for each 1D peak [58].
To increase analysis speed in the second dimension, several approaches have been successfully implemented:
- Monolithic columns offer short regeneration characteristics and high permeability for operation at high flow rates [58]
- Columns with reduced particle sizes, including partially or superficially porous stationary phases or sub-2-μm particles, though these may require UHPLC instrumentation [58]
- Elevated temperatures with ultra-fast gradients to reduce mobile phase viscosity and allow increased flow rates [58]
Diagram 1: Comprehensive 2D-LC System Workflow with Orthogonal Detection
Orthogonal Separation Mode Combinations
The selection of orthogonal separation mechanisms is fundamental to successful 2D-LC implementation. Different combination strategies offer distinct advantages and challenges:
Reversed-phase × Reversed-phase (RP×RP) combinations can provide sufficient orthogonality when different stationary phase chemistries are employed (e.g, C18 in the first dimension and phenylhexyl or pentafluorophenyl in the second dimension) [58]. However, mobile phase compatibility must be carefully considered, as instability can occur when a fluid of low viscosity displaces and penetrates a high viscosity solvent (viscous fingering) [58].
Normal-phase × Reversed-phase (NP×RP) combinations offer high orthogonality but present significant solvent compatibility challenges [58]. The typically apolar normal-phase solvents may not be completely miscible with the aqueous reversed-phase mobile phases, potentially deteriorating the separation and leading to signal interferences [58]. Solutions to this challenge include using micro-flow rates in the first dimension to reduce dilution and solvent volume transferred, or implementing a vacuum evaporation interface to remove incompatible solvents [58].
Hydrophilic interaction liquid chromatography × Reversed-phase (HILIC×RP) has emerged as a particularly promising combination [58]. In this configuration, the mobile phase used in the 1D typically has higher elution strength than that used in the 2D, making micro-flow rates in the first dimension beneficial for reducing dilution and providing flow rates compatible with 2D injection volumes [58].
Table 2: Orthogonal Separation Mode Combinations in 2D-LC
| Combination | Orthogonality | Solvent Compatibility | Best Applications |
|---|---|---|---|
| RP × RP (different chemistries) | Moderate | High | Complex mixtures with diverse hydrophobicity |
| NP × RP | High | Low (requires special interfaces) | Compounds with wide polarity range |
| HILIC × RP | High | Moderate to High | Polar and semi-polar compounds |
| SEC × RP | High | Moderate | Macromolecules and aggregates |
| IEX × RP | High | Moderate | Charged molecules, biologics |
Spiking Studies for Specificity Demonstration
Experimental Design and Protocol
Spiking studies provide a direct and compelling approach to demonstrate method specificity by challenging the chromatographic method with known impurities and degradation products. The protocol involves intentionally spiking the drug substance or drug product with known impurities or degradants at appropriate levels to simulate potential real-world scenarios.
Sample Preparation Protocol:
- Prepare a stock solution of the active pharmaceutical ingredient (API) at the target test concentration
- Prepare separate stock solutions of available impurities and degradants, typically at concentrations representing 0.1-1.0% of the API concentration unless specified otherwise by regulatory thresholds
- Prepare spiked samples by combining:
- 1.0 mL API stock solution
- Appropriate volume of impurity/degradant stock solutions to achieve target levels
- Diluent to final volume
- Prepare control samples without spikes using the same dilution scheme
- Analyze all samples in triplicate using the developed chromatographic method
For forced degradation studies, spike known degradation products (when available) into stressed samples to verify separation from the main peak and from each other. When impurities are not available, compare test results to a second well-characterized procedure [1].
Data Interpretation and Acceptance Criteria
The specificity of the method is demonstrated when:
- Resolution between the analyte peak and all potential impurities/degradants is greater than 2.0
- Peak purity for the main analyte, as determined by PDA and/or MS, confirms no co-elution with any impurity
- Mass balance in forced degradation studies approaches 100%, indicating all degradation products are accounted for and resolved
- Accuracy of the analyte measurement in the presence of impurities/degradants remains within method validation criteria (typically ±15% of known value)
For impurity quantification, accuracy is determined by the analysis of samples spiked with known amounts of impurities, with recovery of 80-120% generally considered acceptable for low-level impurities [1].
Integrated Approach: 2D-LC with Spiking Studies
Comprehensive Specificity Assessment Protocol
The integration of 2D-LC with spiking studies represents the most rigorous approach to specificity testing. This protocol combines the separation power of comprehensive 2D-LC with the targeted challenge of spiking studies, all under the orthogonal detection of PDA and MS.
Integrated Experimental Workflow:
- Forced degradation of the API under relevant stress conditions (acid, base, oxidation, thermal, photolytic)
- Spike potential impurities into the stressed samples at appropriate levels
- First dimension separation using optimal conditions for the primary separation mechanism
- Comprehensive transfer of all 1D effluent to the second dimension via interface valve
- Second dimension separation using orthogonal separation mechanism with rapid gradient
- Simultaneous detection with PDA and MS detectors
- Data analysis for peak purity, resolution, and identification
Diagram 2: Integrated Specificity Assessment Using 2D-LC with Spiking Studies
Case Study: Specificity Assessment for Regulatory Submission
A practical application of this integrated approach involves the development of a stability-indicating method for a small molecule API. The following case study outlines the experimental design and key results:
Materials and Methods:
- API: GSK3β inhibitor structural analogue (similar to PDA-66 referenced in literature) [4]
- Stress conditions: 0.1N HCl (48h), 0.1N NaOH (48h), 3% H₂O₂ (24h), thermal (70°C, 72h), photolytic (1.2 million lux hours)
- Spiked impurities: Five known process-related impurities at 0.5% level
- 2D-LC system: Agilent 1290 Infinity II 2D-LC Solution
- First dimension: HILIC (150 mm × 2.1 mm, 1.8 μm)
- Second dimension: RP-C18 (50 mm × 4.6 mm, 1.8 μm)
- Detection: PDA (190-400 nm) and QDa mass detector
Results and Discussion: The integrated approach successfully resolved all known impurities and degradation products from the main API peak. The HILIC × RP configuration provided exceptional orthogonality, with resolution values exceeding 2.5 for all critical pairs. PDA-based peak purity assessment confirmed spectral homogeneity of the main peak across all stress conditions, with purity angles consistently below purity thresholds. MS detection provided additional confirmation through consistent mass spectra across the API peak and positive identification of degradation products through mass fragmentation patterns.
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Orthogonal Specificity Testing
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Reference Standards | API and impurity quantification | Certified reference materials with documented purity |
| Forced Degradation Reagents | Sample challenging for stability | HCl, NaOH, H₂O₂ for stress studies |
| 2D-LC Mobile Phase Additives | Enhanced separation and detection | Ammonium formate/aceteate for MS compatibility |
| Stationary Phases | Orthogonal separation mechanisms | HILIC, RP-C18, PFP, phenyl for different selectivity |
| Sample Preparation Solvents | Extraction and dissolution | High purity solvents matching mobile phase |
| System Suitability Standards | Method performance verification | Compounds to verify resolution, efficiency, retention |
Method Validation Considerations
When implementing the integrated orthogonal approach for specificity testing, several method validation parameters require particular attention:
Accuracy should be established across the method range by comparison of results between the 2D-LC method and a second well-characterized method, or through spiking studies with known impurities [1]. Documentation should include data from a minimum of nine determinations over three concentration levels, reported as percent recovery of the known, added amount [1].
Precision demonstrations should include repeatability (intra-assay) and intermediate precision (inter-day, inter-analyst, inter-instrument) [1]. For comprehensive 2D-LC methods, special attention should be paid to the reproducibility of the modulation process between dimensions.
Specificity should be demonstrated through a combination of approaches:
- Resolution of the two most closely eluted compounds
- PDA-based peak purity assessment across all peaks of interest
- MS confirmation of compound identity and purity
- Orthogonal separation using 2D-LC
- Spiking studies with known impurities and degradants
The validation data should clearly demonstrate that the method can accurately quantify the analyte of interest without interference from impurities, degradants, or matrix components, providing confidence in the stability-indicating capability of the method for regulatory submissions [41] [1].
Within pharmaceutical analysis, specificity is the ability of a method to unequivocally assess the analyte of interest in the presence of other components that may be expected to be present in the sample matrix, such as impurities, degradation products, or excipients [1]. Demonstrating specificity is a regulatory requirement for stability-indicating methods, as it confirms that a method can accurately measure the active pharmaceutical ingredient (API) and its degradants without interference [59]. This article details the procedure for specificity testing, framed within a broader thesis on employing Photodiode Array (PDA) and Mass Spectrometry (MS) detection to provide orthogonal and complementary data for definitive method validation [15] [3].
Specificity Validation Case Studies
The following case studies illustrate the application of specificity validation for different drug substances and products, highlighting the combined use of PDA and mass spectrometry.
Table 1: Specificity Validation Case Studies for Drug Substances and Products
| Drug Product/ Substance | Analytical Technique | Key Specificity Findings | Chromatographic Conditions | Reference |
|---|---|---|---|---|
| Glycerol Phenylbutyrate (Ravicti) [15] | LC-PDA & LC-MS/MS | Method separated and characterized a novel degradation product formed under acid, alkali, and oxidative forced degradation conditions. | Column: Ascentis Express F5 (2.7 µm, 100 x 4.6 mm)Mobile Phase: 1mM Ammonium Acetate Buffer:ACN (25:75, v/v)Flow Rate: 0.5 mL/min | [15] |
| Rivaroxaban [3] | LC-PDA-ESI-MS/MS | Method separated and characterized three degradation products (DP-1, DP-2, DP-3) and one process-related impurity; specificity established against all known interferents. | Column: Kinetex C18 (150 x 4.6 mm, 5 µm)Mobile Phase: 20mM Ammonium Acetate:Acetonitrile (65:35, v/v)Flow Rate: 1.0 mL/min | [3] |
| Jwagwieum (Herbal Prescription) [60] | HPLC-PDA | Method demonstrated specificity by resolving nine marker compounds (e.g., gallic acid, loganin, glycyrrhizin) in a complex mixture of six herbal medicines. | Column: SunFire C18 (250 x 4.6 mm, 5 µm)Mobile Phase: Water (0.1% Formic Acid) / Acetonitrile GradientFlow Rate: 1.0 mL/min | [60] |
Experimental Protocols for Specificity Testing
A robust specificity validation protocol must demonstrate that the analytical method is unaffected by the presence of interfering peaks at the retention times of the analytes of interest.
Forced Degradation Studies
Forced degradation (stress testing) is performed to validate that the method is stability-indicating and can effectively separate degradation products from the main API [15] [59].
- Stress Conditions: The drug substance or product is subjected to hydrolytic (acidic, basic, neutral), oxidative, thermal, and photolytic stress conditions according to ICH guidelines Q1A(R2) [15] [3].
- Sample Preparation: Stressed samples are analyzed alongside untreated samples and method blanks. For drug products, a placebo mixture (containing all excipients but no API) is also analyzed to exclude excipient interference [59].
- Acceptance Criteria: The API peak should be resolved from all degradation peaks, typically with a resolution (Rs) greater than 1.5, and demonstrate peak purity via PDA and/or MS [59].
Peak Purity Assessment with PDA
A Photodiode Array detector is a powerful tool for assessing peak purity by collecting full spectra across the entire chromatographic peak [1].
- Protocol: The PDA detector is set to collect spectra from 190 nm to 380 nm (or a suitable range) at every data point across a peak [15] [1].
- Analysis: Peak purity is assessed by comparing spectra at the peak's upslope, apex, and downslope. A pure peak will have a high purity match factor, indicating all spectra are homogeneous [1].
- Limitations: PDA can be limited by a lack of UV response, system noise, and similar spectra or low concentrations of co-eluting compounds [1].
Orthogonal Confirmation with Mass Spectrometry
Mass spectrometry provides unequivocal peak purity and identity confirmation, overcoming many of the limitations of PDA detection [1].
- Protocol: MS detection is used in tandem with PDA. The MS is typically operated in full-scan mode to gather data for all ions within a specified mass range (e.g., m/z 100-800) [15].
- Analysis: The extracted ion chromatograms (XICs) for the specific mass-to-charge ratios of the API and potential degradants are examined. Co-elution is confirmed if multiple ions are observed at the same retention time. Tandem mass spectrometry (MS/MS) provides fragmentation patterns that are used to elucidate the structures of degradation products [15] [3].
- Orthogonal Method: An MS-compatible method using a different separation mechanism (e.g., different column chemistry or mobile phase) can serve as a second, orthogonal method to confirm the results of the primary HPLC method [59].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions and Materials for Specificity Validation
| Item Name | Function / Purpose | Examples / Specifications |
|---|---|---|
| Core-Shell Particle Column | Provides high-efficiency separation for resolving complex mixtures of APIs and degradants. | Ascentis Express F5, Kinetex C18 [15] [3] |
| LC-MS Grade Solvents | High-purity solvents to minimize background noise and ion suppression in MS detection. | Acetonitrile (J.T. Baker), Ammonium Acetate (Fisher Chemicals) [15] |
| Forced Degradation Reagents | To intentionally degrade the sample and generate potential degradants for specificity testing. | Hydrochloric Acid, Sodium Hydroxide, Hydrogen Peroxide [15] |
| Reference Standards | Highly characterized materials used to identify analytes and confirm retention times. | Drug Substance Standard, Available Impurity/Degradant Standards [59] |
| Placebo Formulation | A mixture of all excipients without the API; critical for demonstrating no interference in drug product methods. | Prepared according to the drug product's composition [59] |
Signaling Pathways and Workflows
The following diagrams, created with DOT language and compliant with the specified color and contrast rules, illustrate the logical workflow for specificity validation.
Specificity Validation Workflow
Peak Purity Assessment Logic
Leveraging PDA-MS Synergy for Comprehensive Impurity Profiling
The comprehensive profiling of impurities and degradation products is a critical requirement in pharmaceutical development, directly impacting drug safety and efficacy. Regulatory guidelines, such as the ICH Q6A, emphasize the establishment of scientifically justified specifications and validated test procedures for new drug substances and products [61]. This application note details a synergistic analytical approach that combines the complementary strengths of Photodiode Array (PDA) detection and Mass Spectrometry (MS). While MS provides unparalleled sensitivity and structural information based on molecular mass and fragmentation patterns, PDA detection contributes robust spectral data for distinguishing positional isomers, assessing peak purity, and deconvoluting co-eluting peaks [62] [23]. The integrated PDA-MS system offers a powerful, orthogonal platform for achieving definitive impurity identification and characterization, fulfilling rigorous regulatory requirements for specificity testing [61].
The PDA-MS Synergy: Orthogonal Data for Definitive Identification
The combination of PDA and MS detectors creates an analytical system where the whole is greater than the sum of its parts. This synergy is foundational for comprehensive impurity profiling.
PDA Detection Contributions: A PDA detector collects full ultraviolet-visible (UV-Vis) spectra (typically 190–800 nm) for each analyte in real-time across the chromatographic run [62] [23]. This capability provides several critical functions:
- Peak Purity Assessment: By comparing spectra across different points of a chromatographic peak, analysts can determine if a peak arises from a single, pure compound or multiple co-eluting substances [23].
- Isomer Differentiation: Positional isomers, which often yield identical mass spectra and similar retention times, can frequently be distinguished by their distinct UV spectral profiles [62]. This is a notable advantage over MS alone.
- Spectral Deconvolution: Advanced software functions, such as the i-PDeA algorithm, utilize the unique spectral signatures from the PDA to mathematically resolve and quantify co-eluting compounds without physical chromatographic separation [23].
MS Detection Contributions: A mass spectrometer serves as a highly sensitive and selective detector.
- Accurate Mass Measurement: Provides definitive molecular weight and enables elemental composition determination, which is crucial for identifying unknown impurities [63].
- Structural Elucidation: Tandem mass spectrometry (MS/MS) generates fragmentation patterns that offer insights into the molecular structure of impurities [15] [64].
When used together, the UV spectrum from the PDA and the mass data from the MS provide two independent lines of evidence, significantly increasing confidence in impurity identity. The retention time, UV spectrum, accurate mass, and fragmentation pattern together form a powerful multi-parameter identification system [63].
Experimental Protocol: LC-PDA-MS for Impurity Profiling
This section provides a detailed methodology for setting up and applying an LC-PDA-MS system for the analysis of impurities and degradation products.
Instrumentation and Research Reagent Solutions
Table 1: Essential Research Reagent Solutions and Instrumentation
| Item | Function/Description | Example from Literature |
|---|---|---|
| Liquid Chromatograph | Performs the separation of the drug substance from its impurities and degradation products. | UHPLC/UPLC systems for high-resolution, fast separations [62]. |
| PDA Detector | Detects absorbance across a wide UV-Vis range, providing spectral data for peak purity and identity. | SPD-M20A PDA Detector (Shimadzu); collects data from 190–380 nm [15] [63]. |
| Mass Spectrometer | Provides accurate mass and structural information for definitive identification. | Q/TOF-MS for accurate mass; Ion-Trap MS/MS for fragmentation [15] [63]. |
| Analytical Column | Stationary phase for chromatographic separation. | Core–shell particle column (e.g., Ascentis Express F5, 2.7 µm, 100 x 4.6 mm) [15]. |
| Mobile Phase Buffers | Liquid phase for eluting analytes from the column. | 1 mM Ammonium Acetate buffer; Acetonitrile (LC-MS grade) [15]. |
| Reference Standards | Authentic substances for method development and compound confirmation. | Pharmacopeial standards or certified reference materials. |
Detailed Chromatographic and Detection Conditions
The following protocol, adapted from a validated method for glycerol phenylbutyrate, can be optimized for specific drug substances [15].
Chromatographic Conditions:
- Column: Ascentis Express F5 (2.7 µm, 100 x 4.6 mm I.D.) or equivalent.
- Mobile Phase: 1 mM Ammonium Acetate Buffer (pH ≈5.3) : Acetonitrile (25:75, v/v).
- Flow Rate: 0.5 mL/min.
- Injection Volume: 1.0 µL.
- Column Temperature: 40 °C.
- Autosampler Temperature: 15 °C.
PDA Detection Parameters:
- Primary Wavelength: Monitor at the λ-max of the active pharmaceutical ingredient (e.g., 200 nm).
- Spectral Range: 190–380 nm.
- Spectral Sampling Rate: 1.5625 Hz (or higher for fast peaks).
- Peak Purity Analysis: Enabled using instrument software.
Mass Spectrometric Parameters (ESI Positive/Negative Mode):
- Mass Range: m/z 100–800.
- Nebulizing Gas (N₂) Flow: 3.0 L/min.
- Drying Gas (N₂) Flow: 15 L/min.
- Heating Block Temperature: 450 °C.
- Interface Temperature: 250 °C.
- Data-Dependent MS/MS: Enabled to automatically fragment top ions.
Sample Preparation and Forced Degradation
- Stock Solution: Prepare a solution of the drug substance in a suitable solvent (e.g., 30% methanol with 1% acetic acid) at a known concentration [63].
- Forced Degradation (Stress Testing): Subject the drug substance to various stress conditions as per ICH Q1A(R2) guidelines [15]:
- Acidic Hydrolysis: Treat with 0.1 M HCl at room temperature for 1–24 hours.
- Alkaline Hydrolysis: Treat with 0.1 M NaOH at room temperature for 1–24 hours.
- Oxidative Degradation: Treat with 3% H₂O₂ at room temperature for 1–24 hours.
- Quenching and Dilution: Neutralize the stress samples and dilute to an appropriate concentration with the mobile phase or a compatible solvent before injection.
Data Analysis and Workflow
The power of the PDA-MS system is fully realized during data processing and interpretation. The following workflow and corresponding diagram outline the integrated strategy for impurity profiling.
PDA-MS Impurity Profiling Workflow
- Chromatographic Analysis: Inject stress-testing samples and acquire data simultaneously on both PDA and MS detectors.
- Peak Purity Assessment: Using the PDA data, analyze the main peak and all impurity peaks for purity. The software compares spectra across the peak (up-slope, apex, down-slope).
- Spectral Deconvolution: If a peak fails the purity assessment (suggesting co-elution), apply deconvolution algorithms. Tools like i-PDeA use the unique UV spectra of the components to virtually resolve the overlapping peaks and provide individual spectra and quantitative data for each [23].
- MS Data Interpretation: For each chromatographic peak (including deconvoluted components), use the accurate mass to propose elemental compositions. Analyze MS/MS spectra to derive structural information.
- Orthogonal Confirmation: Correlate the UV spectral profile from the PDA with the structural information from the MS. For example, a compound showing a UV spectrum characteristic of a phenolic structure and an [M+H]+ ion consistent with a hydroxylated derivative of the drug provides robust confirmation [62] [63].
- Identification and Reporting: Compile all data—retention time, UV spectrum, accurate mass, and fragmentation pattern—to propose identities for impurities and degradation products.
Table 2: Quantitative Performance Data for a Representative LC-PDA-MS Method [15]
| Parameter | Bulk Drug Substance (LC-PDA) | Pharmaceutical Formulation (LC-MS/MS) |
|---|---|---|
| Linearity Range | 1.40 – 55.84 µg/mL | 2.79 – 111.68 µg/mL |
| Recovery in Plasma | 94.27% | 98.20% |
| System Backpressure | ~67 bar | - |
The integrated LC-PDA-MS platform is a cornerstone technique for modern pharmaceutical analysis, providing an unmatched level of specificity for impurity profiling. By concurrently delivering rich UV spectral data for differentiation and peak purity assessment, and precise mass data for structural elucidation, it directly addresses the core requirements of regulatory specificity testing as outlined in ICH Q6A [61]. The detailed protocols and workflows provided herein empower researchers, scientists, and drug development professionals to implement this powerful technique, thereby enhancing the robustness of their analytical methods and ensuring the safety and quality of pharmaceutical products.
Conclusion
Specificity testing using PDA and MS represents a critical pillar of analytical method validation, ensuring accurate quantification and reliable stability assessment of pharmaceutical compounds. While PDA offers efficient spectral homogeneity evaluation and MS provides definitive mass-based confirmation, their integrated application delivers the most robust specificity demonstration. The evolving regulatory landscape and increasing molecular complexity demand scientifically sound approaches that recognize the limitations of individual techniques. Future directions include advanced data processing algorithms, increased MS accessibility, and standardized practices for orthogonal method integration. By adopting these comprehensive specificity testing strategies, researchers can generate highly reliable analytical data that supports drug development, quality control, and regulatory compliance with greater scientific confidence.
References
- 1. Analytical Method Validation: Back to Basics, Part II [https://www.chromatographyonline.com/view/analytical-method-validation-back-basics-part-ii]
- 2. Method Validation in Regulated Lab presentation | free to view [https://www.powershow.com/view1/c17e6-ZDc1Z/Method_Validation_in_Regulated_Lab_powerpoint_ppt_presentation]
- 3. Development and validation of a stability indicating LC ... [https://pubs.rsc.org/en/content/articlelanding/2014/ra/c4ra00744a]
- 4. A Simple LC-MS/MS Method for the Quantification of PDA- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8840185/]
- 5. Development and validation of stability indicating HPLC–PDA ... [https://fjps.springeropen.com/articles/10.1186/s43094-023-00473-5]
- 6. Development and Validation of a New Eco-Friendly HPLC ... [https://www.mdpi.com/1648-9144/60/10/1686]
- 7. ICH Q2(R2) Validation of analytical procedures [https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline]
- 8. FDA Announces Plan to Phase Out Animal Testing ... [https://www.fda.gov/news-events/press-announcements/fda-announces-plan-phase-out-animal-testing-requirement-monoclonal-antibodies-and-other-drugs]
- 9. Q1 Stability Testing of Drug Substances and Drug Products [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q1-stability-testing-drug-substances-and-drug-products]
- 10. Analytical Method Validation: Key Parameters for ... [https://www.linkedin.com/posts/tonmoy-sarker-tomal_analytical-method-validation-updates-2025-activity-7375894681567760384-8JR5]
- 11. Optimization, Validation and Application of HPLC-PDA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8001753/]
- 12. Validation challenges in liquid chromatography-tandem ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814619307319]
- 13. Pharmaceutical Quality Control Labs (7/93) [https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/pharmaceutical-quality-control-labs-793]
- 14. How to be prepared for Pharmaceutical Validation in 2025 [https://www.gillsprocess.com/blog-1/2024/12/11/how-to-be-prepared-for-pharmaceutical-validation-in-2025]
- 15. LC-MS/MS and LC-PDA Methods for Robust Determination of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12060053/]
- 16. Development of an HPLC-PDA Method for the ... [https://www.sciencedirect.com/science/article/pii/S0362028X24000632]
- 17. Selected new approaches and future perspectives in liquid ... [https://www.sciencedirect.com/science/article/pii/S0928098725001009]
- 18. Peak Spectral Homogeneity in LC/DAD [https://www.mdpi.com/2227-9717/12/12/2887]
- 19. LC–PDA–MS/MS-Based Dereplication Guided Isolation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9404496/]
- 20. Considerations for Collecting Data Suitable for Peak Purity ... [https://support.waters.com/KB_Inf/Empower_Tips_of_the_Week/WKB222744_Considerations_for_Collecting_Data_Suitable_for_Peak_Purity_Determination]
- 21. Peak Purity in Liquid Chromatography, Part I [https://www.chromatographyonline.com/view/peak-purity-liquid-chromatography-part-i-basic-concepts-commercial-software-and-limitations]
- 22. Peak Purity Parameters for PDA Data - Tip255 - Waters [https://support.waters.com/KB_Inf/Empower_Tips_of_the_Week/WKB224471_Peak_Purity_Parameters_for_PDA_Data]
- 23. UV vs Diode-Array ( PDA ) Detectors for (U) HPLC [https://www.ssi.shimadzu.com/service-support/faq/liquid-chromatography/knowledge-base/uv-vs-pda-detectors/index.html]
- 24. Testing of complementarity of PDA and MS detectors using ... [https://pubmed.ncbi.nlm.nih.gov/26753972/]
- 25. New HPLC, MS, and CDS Products from 2024–2025 [https://www.chromatographyonline.com/view/new-hplc-ms-and-cds-products-from-2024-2025-a-brief-review]
- 26. Enhancing Chimeric Fragmentation Spectra Deconvolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12620602/]
- 27. Development of forced degradation and stability indicating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5761119/]
- 28. Forced Degradation Studies: Regulatory Considerations ... [https://www.biopharminternational.com/view/forced-degradation-studies-regulatory-considerations-and-implementation]
- 29. Utilizing tables, figures, charts and graphs to enhance the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10394528/]
- 30. Advanced high-resolution chromatographic strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11576662/]
- 31. Optimization of Detector Parameters to Improve Sensitivity ... [https://www.waters.com/nextgen/ie/en/library/application-notes/2025/optimization-of-detector-parameters-to-improve-sensitivity-using-the-alliance-is-hplc-system-with-pda-detector.html?srsltid=AfmBOorlUqwp4BL_Twh_sERVIa1nOv6nBd5X8gpU8GMgZL4yWFgpzQKR]
- 32. Hot Topics in Mass Spectrometry (Sept 2025) [https://www.chromatographyonline.com/view/hot-topics-in-mass-spectrometry-sept-2025]
- 33. Empower 3 PDA: What is Purity 1 Angle, Purity 1 Threshold ... [https://support.waters.com/KB_Inf/Empower_Breeze/WKB85832_Empower_3_PDA_What_is_Purity_1_Angle_Purity_1_Threshold_How_is_the_peak_purity_determined]
- 34. Calculation of Peak Purity in HPLC - Industrial Pharmacist [https://industrialpharmacist.com/2025/01/calculation-of-peak-purity-in-hplc/]
- 35. Development of a serum protein biomarker panel for the ... [https://www.nature.com/articles/s41598-025-19631-1]
- 36. Artificial Intelligence-Augmented Imaging for Early ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12187166/]
- 37. Informatics strategies for early detection and risk mitigation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11763847/]
- 38. Harnessing Mass Spectrometry for Biomarker Discovery in ... [https://blog.crownbio.com/biomarker-and-mass-spectrometry-aml]
- 39. PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12117729/]
- 40. Scheduled data-independent acquisition mass ... [https://www.sciencedirect.com/science/article/abs/pii/S1387380625001010]
- 41. Liquid Chromatographic Peak Purity Assessments in ... [https://www.chromatographyonline.com/view/liquid-chromatographic-peak-purity-assessments-in-forced-degradation-studies---an-industry-perspective]
- 42. Ultraviolet Detectors: Perspectives, Principles, and Practices [https://www.chromatographyonline.com/view/ultraviolet-detectors-perspectives-principles-and-practices]
- 43. Separation of Chromatographic Co-Eluted Compounds by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8065729/]
- 44. LC-MS/MS Quantitative Assays [https://mscore.web.unc.edu/lc-ms-ms-quantitative-assays/]
- 45. Calibration Practices in Clinical Mass Spectrometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC9467832/]
- 46. Research A Comparative Analysis of Data Analysis Tools ... [https://www.sciencedirect.com/science/article/pii/S1535947623001342]
- 47. PDA baseline noise (Guided Troubleshooting) - WKB97186 [https://support.waters.com/KB_Inst/Chromatography/WKB97186_PDA_Baseline_Noise_Guided_Troubleshooting]
- 48. Impact of Mobile Phase Additives on LC-MS Sensitivity ... [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/analytical-chemistry/small-molecule-hplc/lcms-spice-cannabinoids?srsltid=AfmBOopXbzgHpq4UfRzsiBoGDSMQpY_ss4FfzS5lbm2pSsI1mTROw8Gm]
- 49. Development and Validation of an HPLC-PDA Method for ... [https://www.mdpi.com/2076-3417/15/19/10797]
- 50. The curious case of chlorophyll removal [https://www.sciencedirect.com/science/article/abs/pii/S0308814614006712]
- 51. Peak Purity in HPLC: Assessment, Troubleshooting, and ... [https://www.sepscience.com/peak-purity-in-hplc-assessment-troubleshooting-and-best-practices-11506]
- 52. Optimization of Detector Parameters to Improve Sensitivity ... [https://www.waters.com/nextgen/ie/en/library/application-notes/2025/optimization-of-detector-parameters-to-improve-sensitivity-using-the-alliance-is-hplc-system-with-pda-detector.html?srsltid=AfmBOoqZ0LRs1n8sZcnzL5MKjAe7A65OwLJ76PsKzehJ72ieYLKtS3nW]
- 53. HPLC-PDA-MS/MS as a strategy to characterize and quantify ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7473366/]
- 54. Enhancing chromatographic separation and extraction ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25036197]
- 55. Comparison of high-performance liquid chromatography/ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3833067/]
- 56. Which LC Detector Should I Buy? [https://www.waters.com/blog/which-lc-detector-should-i-buy/?srsltid=AfmBOoq2BgawSiFMKs20kv42XLMIGQDMCPCnVYXilk2P18O-8WbWcuss]
- 57. Qualitative and quantitative analysis data of the major ... [https://www.sciencedirect.com/science/article/pii/S2352340916303109]
- 58. Combining Orthogonal Comprehensive Two-Dimensional ... [https://www.chromatographyonline.com/view/combining-orthogonal-comprehensive-two-dimensional-liquid-chromatography-methods-food-analysis]
- 59. Validation of Stability-Indicating HPLC Methods for ... [https://www.chromatographyonline.com/view/validation-of-stability-indicating-hplc-methods-for-pharmaceuticals-overview-methodologies-and-case-studies]
- 60. Development and Validation of an HPLC–PDA Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12030021/]
- 61. Q6A Specifications: Test Procedures and Acceptance ... [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q6a-specifications-test-procedures-and-acceptance-criteria-new-drug-substances-and-new-drug-products]
- 62. Applicability of liquid and supercritical fluid ... [https://www.sciencedirect.com/science/article/abs/pii/S2468170921000552]
- 63. of ultra performance liquid chromatography-photodiode... Application [https://link.springer.com/article/10.1007/s00217-013-1925-x]
- 64. NP3 MS Workflow: An Open-Source Software System to ... [https://pubmed.ncbi.nlm.nih.gov/38702053/]