Specificity vs Selectivity in Analytical Method Validation: Key Concepts for Reliable Pharmaceutical Analysis
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on distinguishing between specificity and selectivity in analytical method validation.
Specificity vs Selectivity in Analytical Method Validation: Key Concepts for Reliable Pharmaceutical Analysis
Abstract
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on distinguishing between specificity and selectivity in analytical method validation. It clarifies the foundational definitions as per ICH and other regulatory guidelines, explores practical methodologies for assessment, addresses common troubleshooting scenarios, and outlines the requirements for successful method validation. By synthesizing regulatory standards with practical applications, this resource aims to enhance the reliability, accuracy, and regulatory compliance of analytical data in pharmaceutical and bioanalytical workflows.
Demystifying the Definitions: Specificity and Selectivity in Regulatory Contexts
In the realm of analytical method validation, specificity stands as a fundamental parameter, ensuring the reliability and accuracy of data generated in drug development and quality control. Within the broader research context of specificity versus selectivity, it is critical to establish precise, unambiguous definitions. According to the International Council for Harmonisation (ICH) guideline Q2(R1), the core definition of specificity is unequivocal: "Specificity is the ability to assess unequivocally the analyte in the presence of components which may be expected to be present" [1] [2] [3].
The term "unequivocal" itself means unambiguous, clear, and having only one possible meaning or interpretation [4]. This definition underscores that a specific analytical method can accurately identify and quantify the target analyte amidst a sample matrix that typically contains other constituents, such as impurities, degradants, or excipients [1] [3]. It is the guarantee that the measured signal is derived solely from the analyte of interest, free from interference.
Specificity vs. Selectivity: A Critical Distinction
While often used interchangeably, specificity and selectivity represent distinct concepts in analytical chemistry.
- Specificity is the ideal term for methods that respond exclusively to one single analyte. It implies that the method can confirm the identity of a known analyte within a mixture without needing to identify the other components present [1]. Using a common analogy, if you have a bunch of keys, a specific method can identify the one key that opens the lock, without requiring knowledge of the other keys in the bunch [1].
- Selectivity, on the other hand, is a term more often applied to methods that can simultaneously differentiate and quantify multiple different analytes in a single sample [1]. Extending the analogy, a selective method would be able to identify all the keys in the bunch, not just the one that fits the lock [1]. The IUPAC recommends the use of "selectivity" for analytical methods in a broader context [1].
For the purposes of this whitepaper, focused on the core definition, the discussion will center on specificity as defined by ICH.
Experimental Protocols for Demonstrating Specificity
Demonstrating specificity is a procedural cornerstone of method validation. The following detailed protocols outline the key experiments required to prove a method can assess the analyte unequivocally.
Protocol for Forced Degradation Studies
Forced degradation studies, also known as stress testing, are critical for demonstrating specificity by showing that the method can accurately measure the analyte even when decomposition products are present.
- Objective: To prove the method's stability-indicating property by separating the analyte from its degradation products.
- Materials:
- Purified analyte (drug substance)
- Relevant stress agents: Acid (e.g., 0.1M HCl), Base (e.g., 0.1M NaOH), Oxidizing agent (e.g., 3% H₂O₂), Thermal stress (e.g., oven at 70°C), Photolytic stress (e.g., UV light chamber)
- Appropriate analytical instrument (e.g., HPLC system with a DAD or MS detector)
- Methodology:
- Sample Preparation: Subject separate portions of the analyte to various stress conditions to induce approximately 5-20% degradation. Include an unstressed control sample.
- Acid/Base Hydrolysis: Treat the analyte with acid and base at elevated temperatures (e.g., 60°C) for a defined period (e.g., 1-24 hours). Neutralize before analysis.
- Oxidative Degradation: Expose the analyte to an oxidizing agent at room temperature for a set duration.
- Thermal Degradation: Heat the solid analyte in an oven at a specified temperature.
- Photolytic Degradation: Expose the analyte to UV and/or visible light as per ICH Q1B guidelines.
- Analysis: Inject the stressed samples and the control into the analytical system. The chromatogram of the stressed sample should demonstrate baseline resolution (typically Rs ≥ 2.0) between the analyte peak and all degradation peaks [2]. The analyte peak should also be spectrally pure (e.g., confirmed by diode-array detection or mass spectrometry).
Protocol for Interference Testing with Sample Matrix
This protocol verifies that the sample matrix itself does not cause interference at the retention time of the analyte.
- Objective: To confirm that excipients, placebo, or biological matrix components do not co-elute with or obscure the analyte signal.
- Materials:
- Methodology:
- Blank Matrix Analysis: Inject the blank matrix and analyze. The resulting chromatogram should show no peak at the retention time of the analyte [3].
- Standard Analysis: Inject a solution of the analyte standard to confirm its retention time and peak shape.
- Spiked Matrix Analysis: Inject the sample of the blank matrix that has been spiked with a known concentration of the analyte.
- Data Interpretation: The chromatogram from the spiked matrix should show a single, well-defined peak for the analyte. The recovery of the analyte from the spiked matrix should be within acceptable limits (e.g., 98-102%), confirming the matrix does not cause suppression or enhancement of the signal [2]. The resolution between the analyte peak and the closest eluting matrix peak should be sufficient, ideally Rs ≥ 1.7 [2].
Protocol for Critical Peak Separation (Chromatographic Methods)
For chromatographic techniques, specificity is quantitatively demonstrated by the resolution of critical peak pairs.
- Objective: To demonstrate the method's power to separate the analyte from the closest eluting potential interferent.
- Methodology:
- Identify Critical Pair: Prepare a mixture containing the analyte and the component expected to be the most challenging to separate from it (e.g., a structurally similar impurity or a known matrix component).
- Chromatographic Analysis: Inject the mixture and record the chromatogram.
- Calculate Resolution: Determine the resolution (Rs) between the two closest-eluting peaks. The ICH Q2(R1) guideline states that for critical separations, "specificity can be demonstrated by the resolution of the two components which elute closest to each other" [1]. A resolution of Rs ≥ 2.0 is often targeted for baseline separation [2]. The formula for resolution is: Rs = [2(t₂ - t₁)] / (w₁ + w₂) where t is retention time and w is peak width at baseline.
Visualizing Specificity in Analytical Method Validation
The following diagram illustrates the logical workflow and decision points for establishing method specificity, integrating the core protocols.
Specificity Validation Workflow
Quantitative Data and Acceptance Criteria
The demonstration of specificity yields quantitative data that must meet pre-defined acceptance criteria to confirm the method is fit-for-purpose. The table below summarizes the key parameters and their targets.
Table 1: Key Quantitative Parameters for Specificity Assessment
| Parameter | Experimental Approach | Acceptance Criterion | Rationale |
|---|---|---|---|
| Chromatographic Resolution (Rs) [1] [2] | Analysis of a mixture of the analyte and closest-eluting interferent. | Rs ≥ 2.0 (Baseline separation) [2] | Ensures complete separation for accurate integration of analyte and impurity peaks. |
| Peak Purity [2] | Diode-array detection (DAD) or mass spectrometry (MS) of the analyte peak in a stressed sample. | Purity match factor or MS spectrum confirms a single, homogeneous component. | Confirms the analyte peak is not co-eluting with another substance. |
| Analyte Recovery in Matrix [2] | Comparison of analyte response in spiked matrix vs. neat solution. | Typically 98-102% recovery. | Demonstrates the matrix does not suppress or enhance the analyte signal. |
| Blank Matrix Interference [2] [3] | Analysis of blank sample matrix (placebo, untreated plasma, etc.). | No peak at the retention time of the analyte. | Verifies the signal is from the analyte alone. |
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key reagents and materials essential for conducting rigorous specificity experiments.
Table 2: Essential Research Reagent Solutions for Specificity Testing
| Item / Reagent | Function in Specificity Assessment |
|---|---|
| Placebo Formulation / Blank Matrix [2] [3] | Serves as the negative control to test for interference from excipients, buffers, or endogenous components. |
| Forced Degradation Reagents (Acid, Base, Oxidant) [1] [3] | Used to intentionally degrade the analyte, generating impurity and degradation product profiles to challenge the method's separating power. |
| Structurally Related Impurities/Standards | Used to spike into the analyte sample to prove the method can resolve the analyte from known, similar compounds. |
| Chromatographic Column (HPLC/UPLC) | The stationary phase is critical for achieving the necessary separation; robustness testing often involves evaluating columns from different lots or manufacturers [2] [3]. |
| Mass Spectrometry (MS) Detector [2] | Provides definitive confirmation of peak identity and purity, orthogonal to chromatographic retention time. |
In analytical chemistry, selectivity is a fundamental parameter that refers to a method's capability to distinguish and quantify multiple target analytes in a complex mixture without interference from other components in the sample matrix [5] [6]. This concept is often incorrectly used interchangeably with specificity, though they represent distinct methodological attributes. According to IUPAC guidelines, specificity describes the ideal scenario where a method responds exclusively to a single analyte and is considered the ultimate expression of selectivity—a binary property that cannot be graded [5]. In contrast, selectivity is a gradable property that expresses the extent to which a method can determine particular analytes in complex matrices without interference from other components [5].
Within pharmaceutical research and environmental monitoring, establishing method selectivity is crucial for generating reliable data that supports regulatory submissions and ensures product safety [6] [7]. The distinction becomes particularly significant when analyzing complex samples where structurally similar compounds, isomers, impurities, degradants, or matrix components may coexist with the target analytes [8]. A highly selective method can accurately measure each analyte of interest despite these potential interferents, thereby preventing false positives or negatives that could compromise quality control decisions or environmental risk assessments [8].
Theoretical Foundation: The Specificity-Selectivity Distinction
The conceptual relationship between specificity and selectivity represents a critical foundation for understanding analytical method performance. As defined by IUPAC and other scientific organizations, specificity refers to the situation where a method is completely free from interferences and measures only the intended analyte [5]. This represents an absolute characteristic that cannot be graded—methods are either specific or not. In practical analytical chemistry, however, truly specific methods are rare, particularly when working with complex matrices such as biological fluids, environmental samples, or formulated pharmaceutical products [5].
Selectivity, conversely, represents a graduated capability of a method to determine particular analytes in mixtures or matrices without interference from other components [5]. This gradable nature means methods can demonstrate varying degrees of selectivity, from low to high, depending on their ability to distinguish between the target analyte and potential interferents. The relationship between these concepts is hierarchical: specificity represents the ultimate degree of selectivity, where cross-reactivity or interference is reduced to zero [5].
The distinction becomes particularly evident in techniques such as immunological methods, which are sometimes erroneously described as specific. As these methods often demonstrate cross-reactivity with structurally similar compounds, they are more accurately classified as selective rather than specific [5]. This precision in terminology ensures proper methodological characterization and prevents overstatement of analytical capabilities in scientific literature and regulatory submissions.
Quantitative Assessment of Selectivity
Selectivity is evaluated through systematic challenge tests that determine a method's ability to produce accurate results for target analytes despite the presence of potential interferents [6]. The assessment involves demonstrating that measurements of the analytes of interest remain unaffected by other components that are likely to be present in the sample matrix, such as impurities, degradants, excipients, or structurally similar compounds [7].
Experimental Protocols for Selectivity Assessment
For Pharmaceutical Analysis:
- Sample Preparation: Prepare individual solutions of the target analyte, known impurities, degradation products (generated through forced degradation studies), and excipients at expected concentration levels [6] [7].
- Chromatographic Separation: Inject each solution separately into the analytical system (typically HPLC or UHPLC) to determine retention times and peak responses [7].
- Interference Testing: Prepare a mixture containing all components to demonstrate resolution between the analyte peaks and potential interferents. Critical peak pairs should show resolution greater than 1.5 [7].
- Forced Degradation: Subject the analyte to stress conditions (acid/base hydrolysis, oxidation, thermal degradation, photolysis) and demonstrate that degradation products do not interfere with the quantification of the target analyte [6].
For Environmental Analysis (e.g., Pharmaceutical Monitoring in Water):
- Matrix Spiking: Fortify blank water samples (representing different water types: surface water, wastewater, drinking water) with target analytes at relevant concentration levels [9].
- Interference Assessment: Analyze both fortified and unfortified samples to identify potential matrix interferences. In mass spectrometry, monitor for ion suppression or enhancement effects [9].
- Specificity Confirmation: For LC-MS/MS methods, use Multiple Reaction Monitoring (MRM) transitions to confirm analyte identity based on molecular mass and specific fragmentation patterns, ensuring they are distinct from co-eluting matrix components [9].
Table 1: Key Parameters for Selectivity Assessment in Chromatographic Methods
| Parameter | Assessment Method | Acceptance Criteria |
|---|---|---|
| Chromatographic Resolution | Measure separation between analyte and closest eluting potential interferent | Resolution ≥ 1.5 between critical pairs [7] |
| Peak Purity | Use diode array detection or mass spectrometry to evaluate peak homogeneity | Peak purity index ≥ 990 (indicating homogeneous peak) [7] |
| Matrix Effects | Compare analyte response in neat solution versus matrix | Signal suppression/enhancement ≤ 15% [9] |
| Retention Time Stability | Measure consistency of analyte retention times across different conditions | RSD ≤ 1% for retention times [6] |
Methodological Approaches to Generate Selectivity
Different analytical techniques offer varying degrees of inherent selectivity, with methodological choices significantly impacting the ability to distinguish multiple analytes from interferences.
Chromatographic Separation Methods
Chromatographic techniques form the foundation for achieving selectivity in complex mixture analysis through differential partitioning of compounds between stationary and mobile phases [5]. The degree of selectivity depends on the specific interactions between analytes, stationary phase, and mobile phase composition. High-performance liquid chromatography (HPLC) and ultra-high-performance liquid chromatography (UHPLC) achieve selectivity by exploiting differences in analyte polarity, hydrophobicity, ion-exchange properties, or molecular size [7]. Gas chromatography (GC) provides selectivity based on volatility and polarity interactions with the stationary phase [5].
Hyphenated Techniques
The combination of separation techniques with sophisticated detection methods represents a powerful approach to enhance selectivity [5]. Hyphenated techniques such as gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) provide orthogonal selectivity mechanisms by combining physical separation with spectral identification [5] [9]. In these systems, the separation technique resolves analytes in time, while the detection method adds another dimension of selectivity based on mass-to-charge ratios, fragmentation patterns, or spectral signatures [9].
Table 2: Selectivity Comparison Across Analytical Techniques
| Analytical Technique | Selectivity Mechanism | Typical Applications | Selectivity Level |
|---|---|---|---|
| Immunoassays | Antigen-antibody molecular recognition | Clinical diagnostics, biomarker detection | Moderate (subject to cross-reactivity) [5] |
| HPLC with UV Detection | Retention time + spectral information | Pharmaceutical analysis, impurity profiling | Moderate to High [7] |
| GC-MS | Volatility + retention time + mass spectrum | Environmental analysis, volatile organic compounds | High [5] |
| LC-MS/MS (MRM mode) | Retention time + precursor ion + product ion | Trace analysis in complex matrices (e.g., pharmaceuticals in water) | Very High [9] |
| Ion-Selective Electrodes | Molecular recognition at membrane interface | Ion concentration measurement | Low to Moderate (subject to interference) [5] |
Case Study: Selective Pharmaceutical Monitoring in Water
A recent implementation of selective analysis demonstrates the determination of carbamazepine, caffeine, and ibuprofen in water and wastewater using UHPLC-MS/MS [9]. This method exemplifies modern approaches to achieving high selectivity through:
- Chromatographic Separation: UHPLC provides initial selectivity by separating compounds based on hydrophobicity and column chemistry with high efficiency [9].
- Mass Spectrometric Detection: Tandem mass spectrometry in Multiple Reaction Monitoring (MRM) mode adds orthogonal selectivity by monitoring specific precursor-to-product ion transitions for each compound [9].
- Sample Preparation Optimization: Solid-phase extraction selectively concentrates target analytes while reducing matrix interferences without requiring solvent evaporation, aligning with green chemistry principles [9].
The method successfully demonstrated specificity (as defined in ICH guidelines) with correlation coefficients ≥0.999, precision (RSD <5.0%), and accurate recovery rates from 77-160% across the target analytes, highlighting the practical achievement of high selectivity in a complex environmental matrix [9].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Selectivity Experiments
| Reagent/Material | Function in Selectivity Assessment | Application Examples |
|---|---|---|
| Chromatographic Columns | Differential separation of analytes based on chemical properties | C18, phenyl, cyano, HILIC, chiral stationary phases [7] |
| Mass Spectrometry Reference Standards | Method calibration and analyte identification | Certified reference materials for target analytes and internal standards [9] |
| Forced Degradation Reagents | Generation of potential degradants for interference studies | Acid (HCl), base (NaOH), oxidant (H₂O₂), thermal and photolytic stress conditions [6] |
| Sample Preparation Sorbents | Selective extraction and cleanup of target analytes | Solid-phase extraction cartridges (C18, mixed-mode, polymeric) [9] |
| Matrix Components | Challenge testing with potential interferents | Placebo formulations (excipients), biological fluids, environmental matrix samples [6] [7] |
Selectivity represents a fundamental gradable property of analytical methods that enables reliable measurement of multiple analytes in the presence of potential interferents. The precise distinction between selectivity and specificity is crucial for proper method characterization, with specificity representing the ultimate, non-gradable form of selectivity where no interferences occur [5]. Through strategic implementation of chromatographic separation, hyphenated techniques, and appropriate sample preparation, analytical scientists can achieve the necessary selectivity to address complex analytical challenges in pharmaceutical and environmental analysis [5] [9].
The experimental protocols and case studies presented provide a framework for systematically evaluating and demonstrating method selectivity, emphasizing the importance of challenge tests with potential interferents relevant to the sample matrix [6] [7]. As analytical challenges continue to evolve with increasing matrix complexity and lower detection limit requirements, the fundamental role of selectivity in ensuring data quality and reliability remains paramount in analytical method validation.
In the highly regulated world of pharmaceutical development, the validation of analytical methods is a critical prerequisite for ensuring drug safety, efficacy, and quality. Among the various validation parameters, the concepts of specificity and selectivity are fundamental, yet their distinction often creates confusion among even experienced scientists. The International Council for Harmonisation (ICH) guideline Q2(R1) provides definitions, but practical understanding requires clear, relatable illustrations [10]. Within this context, the "bunch of keys" analogy emerges as an exceptionally powerful tool for delineating the precise difference between these two parameters. This whitepaper explores this analogy in depth, framing it within the broader scope of analytical method validation research and providing the experimental protocols necessary for its practical demonstration in a regulatory-compliant laboratory setting.
The consistent mix-up between specificity and selectivity stems not from a lack of technical knowledge, but from the absence of a persistent mental model. Analogies bridge abstract regulatory concepts with tangible, everyday objects, thereby enhancing comprehension and retention. For researchers, scientists, and drug development professionals, a firm grasp of this distinction is not merely academic; it is essential for designing validation protocols, interpreting data correctly, and successfully navigating regulatory audits [1] [11].
Defining the Concepts: Specificity vs. Selectivity
Official Definitions and the Core Distinction
According to the ICH Q2(R1) guideline, specificity is defined as "the ability to assess unequivocally the analyte in the presence of components which may be expected to be present" [1]. In essence, a specific method can accurately identify and/or quantify the target analyte amidst a mixture of potentially interfering substances, such as impurities, degradation products, or sample matrix components. The European guideline on bioanalytical method validation further refines the concept of selectivity, defining it as the ability "to differentiate the analyte(s) of interest and IS from endogenous components in the matrix or other components in the sample" [1].
The fundamental distinction lies in the scope of analysis. A method is specific when it is concerned with a single analyte, ensuring that the measured response is due to that analyte alone. A method is selective when it can successfully identify and/or quantify multiple different analytes within the same sample, distinguishing each one from all others [1] [11]. The International Union of Pure and Applied Chemistry (IUPAC) notes that "specificity is the ultimate of selectivity" and often recommends the use of the term 'selectivity' in analytical chemistry, as very few methods respond to only one analyte [10].
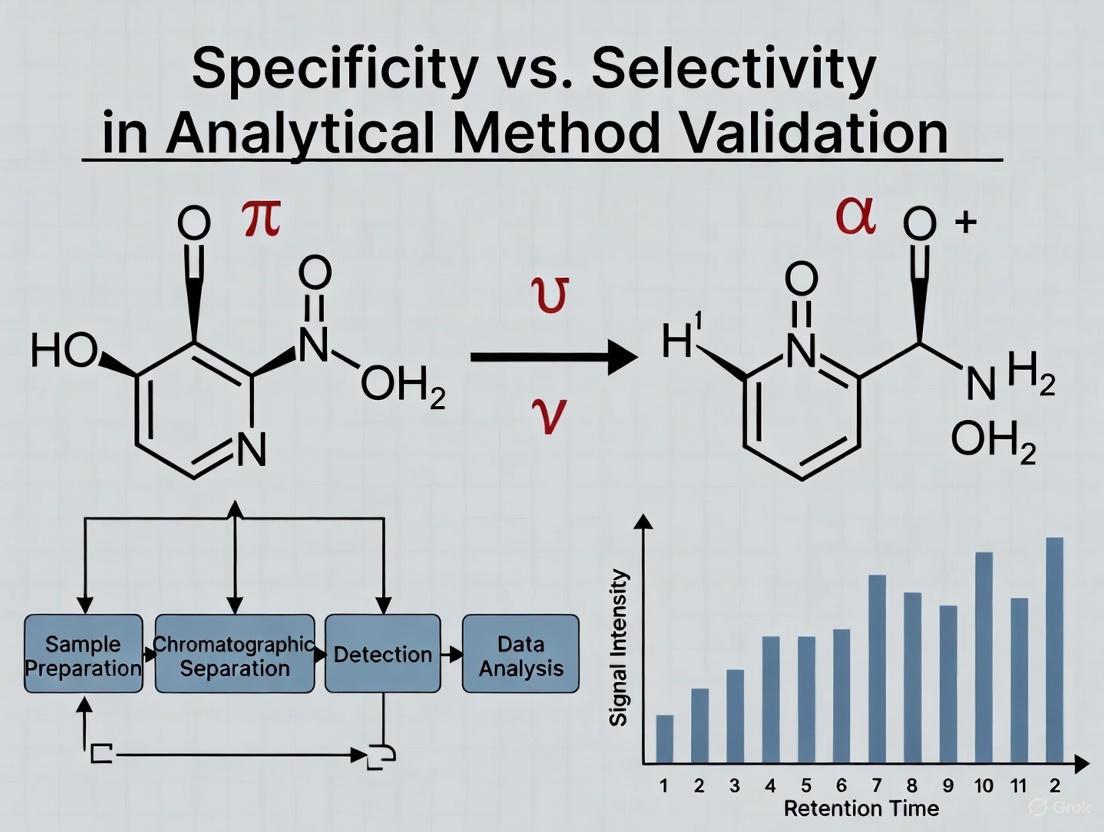
The "Bunch of Keys" Analogy
The "bunch of keys" provides a perfect, intuitive model for understanding this distinction [1] [11].
- The Scenario: Imagine a bunch of keys, where each key is a different chemical entity in a sample mixture. The lock on a specific door represents the detector or the analytical method.
- Illustrating Specificity: In this context, specificity is the ability of the lock to be opened by one, and only one, correct key—the analyte of interest. The goal is not to identify what the other keys are, but simply to ensure that they do not open the lock. The method is specific if it responds only to the target key (analyte) and not to any others (potential interferents) [1].
- Illustrating Selectivity: Selectivity, however, requires the identification and differentiation of all keys in the bunch. It demands that the method can not only identify the one correct key for the lock but also recognize and distinguish all other keys present—be they for a car, a cabinet, or a safe. In analytical terms, a selective method can resolve, identify, and quantify all analytes of interest in a multi-component mixture [1] [11].
The following diagram visualizes this logical relationship, mapping the analogy to the technical parameters and their outcomes.
Regulatory and Experimental Framework
Validation Requirements Across Guidelines
The requirement to demonstrate either specificity or selectivity is mandated by all major regulatory bodies, though the terminology can vary. The following table summarizes the position of key international guidelines, highlighting that while ICH Q2(R1) focuses on "specificity," other frameworks acknowledge both terms or emphasize "selectivity" for multi-analyte methods [10].
Table 1: Regulatory Stance on Specificity and Selectivity in Method Validation
| Regulatory Guideline | Primary Terminology Used | Context and Requirements |
|---|---|---|
| ICH Q2(R1) | Specificity | Required for identification, impurity, and assay tests. For chromatography, critical separation is demonstrated by the resolution of the two closest-eluting components [1] [10]. |
| FDA | Specificity/Selectivity | Acknowledges both terms, requiring demonstration that the method can differentiate the analyte in the presence of other components [10]. |
| European Pharmacopoeia | Specificity | Follows ICH definitions, emphasizing the need to detect the analyte unequivocally among potential interferents [10]. |
| USP | Specificity | Validation parameter includes specificity, with emphasis on peak purity for chromatographic methods [10]. |
Experimental Protocols for Demonstration
Demonstrating specificity and selectivity involves a series of deliberate experiments designed to challenge the method with potential interferents. The specific protocols depend on the type of analytical procedure (e.g., identification, assay, or impurity test).
Protocol for Specificity (Assay and Purity Tests)
This protocol is designed to prove that the assay result for the active ingredient is unaffected by the presence of impurities, degradation products, or excipients [1] [10].
Sample Preparation:
- Pure Analyte (Reference): Prepare a sample of the pure drug substance (analyte) at the target concentration.
- Placebo/Matrix Blank: Prepare a sample containing all excipients or the full sample matrix without the analyte.
- Spiked Mixture: Spike the pure analyte with appropriate levels of all available impurities, degradation products, and excipients. For forced degradation studies, stress the drug product (e.g., with heat, light, acid, base, oxidant) to generate degradation products [1] [12].
Analysis and Acceptance Criteria:
- Analyze all samples using the method.
- The placebo/matrix blank should show no interference, meaning no peak or signal at the retention time/migration position of the analyte.
- The spiked mixture or stressed sample should demonstrate that the analyte peak is pure (e.g., as determined by a Diode Array Detector or Mass Spectrometer) and that the assay result for the analyte is equivalent (within predefined acceptance criteria) to the result obtained from the unspiked pure analyte reference [10].
Protocol for Selectivity (Multi-Analyte Methods)
This protocol is used for methods that quantify multiple analytes simultaneously, such as impurity profiling or bioanalytical methods.
Sample Preparation:
- Individual Analyte Standards: Prepare separate solutions of each individual analyte of interest at the target concentration.
- Mixed Standard: Prepare a solution containing all analytes of interest at their target concentrations.
- Matrix Sample Spiked with Analytes: Spike a representative blank matrix (e.g., plasma, placebo) with the mixture of all analytes.
Analysis and Acceptance Criteria:
- Analyze all samples.
- The method must be able to resolve all analytes from each other in the mixed standard and the spiked matrix sample. For chromatographic methods, the resolution between the pair of analytes that elute closest to each other should be greater than a specified limit (e.g., Rs > 1.5) [1] [10].
- The peak purity of each analyte should be confirmed in the mixed standard.
- The quantitation of each analyte in the mixed standard and the spiked matrix sample should be accurate and precise when compared to the individual standard solutions.
The workflow for these experimental studies, from sample preparation to data interpretation, is outlined in the following diagram.
The Scientist's Toolkit: Essential Materials for Validation
Successfully conducting these experiments requires a set of well-characterized reagents and materials. The following table details the essential components of a "Research Reagent Solution" for specificity/selectivity studies.
Table 2: Key Research Reagents and Materials for Specificity/Selectivity Studies
| Reagent/Material | Function in Validation | Critical Quality Attributes |
|---|---|---|
| Drug Substance (Analyte) Reference Standard | Serves as the primary benchmark for identity, retention time, and response factor. | High purity (>98%), fully characterized structure, known impurities profile. |
| Known Impurity and Degradation Product Standards | Used to spike samples to demonstrate resolution from the main analyte and from each other. | Certified purity and concentration, structural confirmation. |
| Placebo Formulation (for Drug Product) | Represents the sample matrix without the active ingredient to test for interference from excipients. | Representative of the final drug product composition, batch-to-batch consistency. |
| Blank Matrix (e.g., Plasma, Serum) | For bioanalytical methods, used to test for interference from endogenous components. | Sourced from appropriate species, confirmed to be free of analytes. |
| Appropriate Solvents and Mobile Phases | Used for sample preparation, dilution, and as the eluent in chromatographic systems. | HPLC/GC grade, low in UV absorbance, free of particulates. |
| System Suitability Standards | A reference mixture used to verify that the total analytical system is performing adequately before and during the analysis. | Contains key analytes to confirm parameters like resolution, precision, and tailing factor. |
The 'bunch of keys' analogy transcends being a mere memory aid; it provides a robust conceptual framework that aligns perfectly with regulatory definitions and practical laboratory workflows. By internalizing this model, scientists can more effectively design, execute, and interpret the validation studies that are the bedrock of pharmaceutical quality control. As analytical techniques continue to evolve towards the simultaneous analysis of increasingly complex mixtures, the principle of selectivity—the ability to identify every key in the bunch—will only grow in importance. A deep and intuitive understanding of the distinction between specificity and selectivity, therefore, remains an indispensable asset for any professional committed to excellence in drug development and validation research.
Analytical method validation stands as a cornerstone of pharmaceutical development, ensuring the reliability, accuracy, and reproducibility of data supporting drug safety and efficacy. The comparative analysis of validation guidelines issued by major international regulatory bodies reveals a complex landscape of harmonized and divergent requirements. Understanding the nuances between the International Council for Harmonisation (ICH), the United States Food and Drug Administration (FDA), and the European Medicines Agency (EMA) is crucial for global drug development strategies. This technical examination frames these regulatory perspectives within a broader scientific investigation into specificity versus selectivity, fundamental analytical parameters that define a method's ability to measure the analyte accurately amidst interfering components [13].
The regulatory harmonization achieved through ICH provides a foundational framework, while regional implementations by FDA and EMA introduce critical distinctions in application and emphasis. For researchers and drug development professionals, navigating these aligned yet distinct pathways demands both technical precision and strategic regulatory insight. This guide provides a detailed comparison of these frameworks, emphasizing their practical implications for analytical method validation, particularly through the lens of specificity and selectivity requirements [13] [14].
Regulatory Frameworks and Governance
ICH: The Global Standard-Setter
The ICH Q2(R1) guideline, titled "Validation of Analytical Procedures: Text and Methodology," represents the internationally harmonized foundation for analytical method validation. Established through collaboration between regulatory authorities and pharmaceutical industries from the European Union, United States, Japan, and other regions, ICH Q2(R1) unifies principles previously contained in separate Q2A and Q2B documents. This guideline provides the core validation parameters and methodology for experimental data required for registration applications, creating a common scientific language for analytical procedure validation across most major markets [15].
FDA: The Prescriptive Regulator
The FDA's approach to method validation is characterized by a rule-based, prescriptive framework codified primarily in 21 CFR Parts 210 and 211. The FDA emphasizes strict adherence to predefined protocols with detailed requirements for validation data generation and documentation. The agency's current thinking reflects a lifecycle approach to validation, incorporating risk management principles and emphasizing method robustness throughout its application. FDA inspectors focus heavily on data integrity and ALCOA principles (Attributable, Legible, Contemporaneous, Original, Accurate) during audits, with particular attention to documentation traceability and raw data verification [13] [14].
EMA: The Principle-Based Coordinator
The EMA operates as a coordinating network across EU Member States rather than a centralized authority like FDA. Its scientific guidelines, including those for method validation, are compiled in EudraLex Volume 4. The EMA's approach is principle-based and directive, expecting manufacturers to interpret guidelines within a comprehensive quality system framework. Unlike the FDA's prescriptive style, EMA emphasizes risk-based thinking and integrated quality management systems (QMS), requiring more extensive justification of scientific decisions rather than strict protocol adherence. The EMA has recently adopted the ICH M10 guideline for bioanalytical method validation, replacing its previous standalone guidance, demonstrating the ongoing harmonization efforts across regions [16] [14] [17].
Table 1: Fundamental Regulatory Structures and Approaches
| Aspect | ICH | FDA | EMA |
|---|---|---|---|
| Primary Guidance | Q2(R1) Validation of Analytical Procedures | 21 CFR Parts 210/211; Lifecycle Approach | ICH M10 (Bioanalytical); EudraLex Volume 4 |
| Regulatory Style | Scientifically harmonized | Prescriptive, rule-based | Principle-based, quality system focused |
| Geographical Scope | International (EU, US, Japan, etc.) | United States | European Union member states |
| Decision-Making | Consensus-based | Centralized federal authority | Network of national authorities |
| Key Emphasis | Analytical performance parameters | Data integrity, protocol adherence | Risk management, QMS integration |
Core Validation Parameters: Comparative Analysis
Specificity and Selectivity: Foundational Concepts
Within analytical method validation, specificity and selectivity represent complementary parameters addressing a method's ability to measure the analyte unequivocally in the presence of interfering components. While terminology differs slightly between guidelines, the fundamental requirement remains consistent: demonstration that the method can accurately quantify the target analyte despite potential interferents from impurities, degradation products, matrix components, or other analytes.
Specificity is often described as the ultimate expression of selectivity – the ability to measure accurately in the presence of all potentially interfering substances. In chromatographic methods, this typically requires demonstration of peak purity using diode array detection or mass spectrometry, while for spectroscopic methods, the absence of spectral overlaps must be verified. For bioanalytical methods, the EMA (through ICH M10) emphasizes matrix effect evaluation specifically, requiring assessment of ionization suppression or enhancement in mass spectrometry-based methods [16].
Comprehensive Parameter Comparison
The following table provides a detailed comparison of validation parameter requirements across the three regulatory frameworks, highlighting distinctions in emphasis and acceptance criteria that impact method development and validation strategies.
Table 2: Analytical Method Validation Parameters Comparison
| Validation Parameter | ICH Q2(R1) | FDA Approach | EMA/ICH M10 Approach |
|---|---|---|---|
| Specificity/Selectivity | Required; demonstrate unequivocal assessment | Required; forced degradation studies expected | Required; matrix effects assessment for bioanalytical |
| Accuracy | Required; recovery studies 80-120% typically | Required; protocol-specific criteria | Required; may emphasize patient population relevance |
| Precision | Required (repeatability, intermediate precision) | Required; includes system suitability | Required; may require additional ruggedness testing |
| Detection Limit (LOD) | Required for impurity methods | Required when applicable | Required when applicable |
| Quantitation Limit (LOQ) | Required for impurity methods | Required when applicable | Required when applicable |
| Linearity | Required; minimum 5 concentration points | Required; protocol-specific range | Required; may emphasize therapeutic range |
| Range | Required; established from linearity studies | Required; justified based on application | Required; may consider clinical relevance |
| Robustness | Recommended; often tested during development | Expected; system suitability controls | Required; quality by design approach encouraged |
Regulatory Emphasis and Documentation
The regulatory emphasis on certain validation parameters differs between agencies, reflecting their distinct philosophical approaches. The FDA's prescriptive nature manifests in detailed expectations for protocol pre-specification and strict adherence to predefined acceptance criteria. Any deviation triggers rigorous investigation and documentation. FDA submissions require comprehensive raw data presentation with explicit statistical analysis supporting validation conclusions [14].
In contrast, EMA's principle-based approach emphasizes scientific justification behind selected parameters and acceptance criteria. The EMA may place greater emphasis on the clinical relevance of validation results, particularly for bioanalytical methods supporting pharmacokinetic studies. Documentation for EMA submissions must demonstrate how validation parameters ensure patient safety and reliable results within the context of clinical use, with stronger integration into the overall Pharmaceutical Quality System [16] [14].
Experimental Protocols for Specificity and Selectivity Assessment
Chromatographic Method Protocol
For HPLC/UV-DAD methods, the following protocol provides comprehensive specificity/selectivity validation:
Materials and Equipment:
- HPLC system with diode array detector (DAPI)
- Reference standard of analyte (highest available purity)
- Potentially interfering substances (impurities, degradation products, matrix components)
- Appropriate chromatographic column and mobile phase components
Experimental Procedure:
- System Preparation: Equilibrate HPLC system with mobile phase at specified flow rate and column temperature
- Individual Solutions: Prepare separate solutions of analyte and each potential interfering compound at expected concentration levels
- Forced Degradation Samples: Subject analyte to stress conditions (acid/base, oxidation, thermal, photolytic) and analyze degraded samples
- Resolution Solution: Prepare mixture containing analyte and all potential interferents at expected maximum concentrations
- Chromatographic Analysis: Inject individual solutions and mixture using validated method parameters
- Peak Purity Assessment: Use DAD to collect spectra across each peak and verify purity through spectral overlay and match factor calculations
- Resolution Calculation: Measure resolution between analyte peak and closest eluting interferent
Acceptance Criteria:
- Resolution between analyte and all interferents ≥ 2.0
- Peak purity index ≥ 990 (on scale of 0-1000)
- No co-elution observed in mixed standard injection
- Spectral homogeneity confirmed across entire peak
Ligand Binding Assay Protocol
For immunoassay methods requiring selectivity assessment:
Materials and Equipment:
- Reference standard and quality control samples
- Potentially cross-reacting structurally similar compounds
- Target population and normal control matrices
- Required reagents, plates, and detection equipment
Experimental Procedure:
- Preparation of Cross-reactivity Solutions: Spike potentially interfering compounds at 100x expected physiological concentration into analyte solutions at low, medium, and high QC levels
- Matrix Selection: Collect matrices from at least 10 individual sources of relevant population (disease state if applicable)
- Parallelism Assessment: Prepare serial dilutions of analyte in different matrices and compare dose-response curves to reference standard
- Recovery Assessment: Spike known analyte concentrations into different matrices and calculate percentage recovery
- Assay Performance: Analyze all samples in validated assay format
Acceptance Criteria:
- Cross-reactivity with structurally similar compounds < 5%
- Parallelism demonstrated with curves parallel to reference standard
- Mean recovery within 85-115% across all matrices
- No significant matrix effects observed across individual sources
Visualization of Regulatory Relationships
Regulatory Guideline Relationships and Emphases
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Analytical Method Validation
| Reagent/Material | Function in Validation | Specific Application |
|---|---|---|
| Reference Standard | Primary measurement standard | Quantification and method calibration |
| Forced Degradation Reagents | Specificity demonstration | Acid, base, oxidants, heat, light sources |
| Matrix Components | Selectivity assessment | Plasma, serum, tissue homogenates |
| System Suitability Mixtures | System performance verification | Resolution and precision testing |
| Stability Solutions | Method robustness evaluation | Short-term and long-term stability |
| Cross-reactivity Compounds | Specificity confirmation | Structurally similar molecules |
Strategic Implementation and Compliance
Alignment with Specificity vs Selectivity Research
The regulatory perspectives on specificity and selectivity reflect the ongoing scientific discourse around these fundamental analytical concepts. The FDA's emphasis on forced degradation studies aligns with a rigorous approach to specificity verification, demanding demonstration that methods can distinguish the analyte from all potential degradation products. The EMA's focus on matrix effects in bioanalytical methods through ICH M10 represents a selectivity-centered approach, ensuring accurate measurement despite biological matrix variations [16] [14].
Within a broader thesis on specificity versus selectivity, these regulatory distinctions highlight how theoretical concepts manifest in practical requirements. The harmonization through ICH establishes common definitions, while regional implementations reflect different risk tolerance and historical approaches to analytical validation. Understanding these nuances enables development of validation strategies that satisfy both specific technical requirements and overarching regulatory expectations [13] [15].
Strategic Compliance Recommendations
Successful navigation of the FDA and EMA regulatory landscapes requires both technical excellence and strategic planning:
- Adopt ICH Q2(R1) Foundation: Implement ICH Q2(R1) as the core validation framework, then build agency-specific elements upon this foundation
- Document Rationale: Maintain thorough documentation justifying validation approaches, particularly for parameters where guidelines differ or allow flexibility
- Implement Risk Assessment: Apply risk-based principles to validation designs, focusing resources on critical methods with greatest impact on product quality and patient safety
- Prepare Agency-Specific Documentation: Adapt validation summaries to emphasize elements each agency prioritizes – raw data and protocol adherence for FDA, scientific justification and QMS integration for EMA
- Leverage Harmonization: Utilize the convergence between EMA and FDA through adoption of ICH M10 for bioanalytical methods to streamline global development [16]
The evolving regulatory landscape continues to emphasize lifecycle approach to method validation, with increasing harmonization through ICH initiatives. Maintaining awareness of guideline updates and their practical implementation remains essential for successful global regulatory strategy and efficient market access for pharmaceutical products.
In the rigorous world of analytical chemistry and bioanalytical method validation, the precise use of terminology is not merely academic—it forms the bedrock of reproducible science, regulatory compliance, and clear scientific communication. Among the most persistent sources of confusion lies in distinguishing between specificity and selectivity. While often used interchangeably in casual laboratory parlance, these terms carry distinct technical meanings with significant implications for method validation protocols. This whitepaper examines the nuanced relationship between these two fundamental analytical concepts, with a particular focus on the International Union of Pure and Applied Chemistry (IUPAC) recommendations that frame specificity as the ultimate expression of selectivity. Within the context of analytical method validation research, understanding this hierarchy is essential for researchers, scientists, and drug development professionals who must design validation experiments that meet both scientific and regulatory standards.
The debate is not purely semantic; it strikes at the heart of how we characterize a method's ability to measure an analyte accurately within a complex matrix. As per IUPAC's recommendations, the term specificity should describe the ideal, but often theoretically unattainable, scenario where a method responds exclusively to one single analyte. In contrast, selectivity refers to the practical ability of a method to determine several analytes simultaneously in the presence of potential interferents [1] [18]. This paper will explore the technical definitions, practical applications, experimental protocols for demonstration, and the ongoing scientific discourse surrounding these pivotal analytical properties.
Defining the Concepts: IUPAC's Evolving Terminology
The Official Definitions and Historical Context
The IUPAC, as the international authority on chemical nomenclature and terminology, provides the foundational definitions for the analytical sciences [19] [20]. According to IUPAC recommendations, selectivity is defined as the "property of a measuring system, used with a specified measurement procedure, whereby it provides measured quantity values for one or more measurands such that the values of each measurand are independent of other measurands or other quantities in the phenomenon, body, or substance being investigated" [18]. In simpler terms, selectivity is the ability of a method to differentiate and quantify multiple analytes within a complex sample, ensuring that the measurement of each is not skewed by the presence of the others.
Specificity, within this framework, is considered the ultimate degree of selectivity [18]. It represents an ideal scenario where a method is capable of responding to one, and only one, analyte. The IUPAC Compendium of Terminology in Analytical Chemistry (the "Orange Book") serves as the authoritative resource for these definitions, with the latest edition published in 2023 reflecting the ongoing evolution in the field [19]. The historical development of this terminology reveals a gradual shift towards precision, moving away from the interchangeable usage that has long clouded the field.
The Practical Distinction: A Conceptual Analogy
A commonly used analogy effectively illustrates the practical distinction between these concepts:
- Specificity is akin to identifying a single, specific key in a bunch that can open a particular lock. The focus is solely on finding that one correct key; identifying the other keys on the ring is not required [1] [11].
- Selectivity, using the same analogy, requires the identification of all keys in the bunch, not just the one that opens the lock [1] [11].
This analogy clarifies that specificity concerns itself with a single target, while selectivity involves a broader analytical scope, characterizing multiple components within a mixture. In practical analytical chemistry, achieving true specificity is often considered nearly impossible because real-world samples may contain numerous chemicals that could potentially interfere [18]. Therefore, selectivity is the more commonly demonstrated and practical property for most analytical methods.
Regulatory Landscape and Guidelines
ICH vs. IUPAC: A Comparative Analysis
The regulatory landscape for analytical method validation features guidelines that sometimes diverge in their terminology, creating a source of ongoing debate. The International Council for Harmonisation (ICH) guideline Q2(R1), a cornerstone for pharmaceutical analysis, defines specificity as "the ability to assess unequivocally the analyte in the presence of components which may be expected to be present" [1]. This definition, heavily focused on the demonstration of a lack of interference, is the primary term used in the guideline for validation parameters, and it is a required validation parameter for identification tests, impurity tests, and assays [1].
Notably, the term "selectivity" does not appear in ICH Q2(R1), highlighting a fundamental divergence from IUPAC's lexicon. In contrast, other guidelines, such as the European guideline on bioanalytical method validation, do employ the term "selectivity," defining it as the ability "to differentiate the analyte(s) of interest and IS from endogenous components in the matrix or other components in the sample" [1]. This regulatory patchwork means that professionals must be conversant with both sets of terminology, applying the appropriate terms based on the regulatory context of their work.
Table 1: Comparing Analytical Terminology Across Guidelines
| Term | IUPAC Recommendation | ICH Q2(R1) Guideline | Practical Implication |
|---|---|---|---|
| Selectivity | The primary, preferred term. The ability to measure multiple analytes without mutual interference. | Not explicitly mentioned or defined. | A practical, measurable property for multi-analyte methods. |
| Specificity | The ultimate degree of selectivity; an ideal where a method responds to only one analyte. | The key term used; defined as the ability to assess the analyte unequivocally in the presence of expected components. | Often treated as a synonym for selectivity in regulated pharma labs. |
| Philosophy | Views selectivity as a scalable property, with specificity being its absolute, ideal form. | Uses specificity as the catch-all term for a method's ability to distinguish the analyte. | Creates a disconnect between scientific and regulatory language. |
The Case for IUPAC's Preference
IUPAC's preference for "selectivity" as the overarching term is rooted in scientific pragmatism. Given that very few analytical techniques are truly specific to a single analyte in all possible scenarios, selectivity is a more honest and accurate descriptor [18]. It acknowledges that methods can possess varying degrees of ability to distinguish an analyte from interferents. This conceptualization allows for a more granular and quantitative assessment of a method's performance. The recommendation is that the term "specificity" should be reserved for those rare cases where absolute selectivity has been demonstrated, a situation that is more theoretical than practical [18]. This nuanced view encourages a more critical and evidence-based approach to method validation.
Experimental Protocols for Demonstrating Selectivity and Specificity
Core Methodological Principles
Demonstrating selectivity (or specificity, as per ICH) is a fundamental requirement in method validation. The core principle is to provide evidence that the analytical signal attributed to the analyte is unequivocally derived from that analyte and is not significantly influenced by other substances present in the sample [18]. This involves a series of experiments designed to challenge the method with potential interferents.
A method is considered selective when the analytical signal of the analyte can be separated from other signals, and where each signal depends on a specific property of the analyte to be measured [18]. The experimental design must be tailored to the type of method (e.g., chromatographic vs. ligand binding assay) and the nature of the sample matrix.
Detailed Experimental Workflows
The following workflows outline the key experiments required to demonstrate selectivity for different analytical purposes.
Diagram 1: General Selectivity Assessment Workflow
Protocol for Chromatographic Methods (e.g., HPLC, LC-MS)
- Analysis of Blank Matrix: Inject a sample of the blank matrix (e.g., plasma, formulation buffer) to confirm the absence of interfering peaks at the retention times of the analyte and internal standard [1].
- Analysis of Spiked Matrix: Inject a sample of the matrix spiked with the analyte at the target concentration to confirm the analyte's retention time and response.
- Interference Testing: Inject samples containing potential interferents individually. These interferents should include:
- Excipients from the drug formulation.
- Pharmacologically relevant co-administered drugs.
- Known metabolites.
- Degradation products generated from forced degradation studies [1].
- Co-injection: Inject a sample containing both the analyte and the potential interferents to demonstrate that the resolution between the analyte peak and the closest eluting interference meets predefined criteria (e.g., resolution factor Rs > 2.0) [1].
- Forced Degradation Studies: Subject the analyte to stress conditions (e.g., acid, base, oxidative, thermal, photolytic) to generate degradation products. Then, analyze the stressed sample to demonstrate that the analyte peak is pure and free from co-eluting degradants, and that the degradation products are resolved from the analyte and from each other [1].
Protocol for Ligand Binding Assays (LBAs)
- Assessment of Cross-Reactivity: Test the assay against structurally similar compounds, related substances, active forms, and degradation products [18]. The concentrations of these interfering substances should be similar to or higher than their expected physiological concentrations.
- Parallelism Testing: Demonstrate that the measured concentration of the analyte is consistent when the sample is diluted, indicating a lack of matrix interference.
- Spiked Recovery in Different Matrices: Spike the analyte into different lots of the matrix (e.g., multiple lots of human plasma) and measure recovery. The results should be consistent across lots.
Table 2: Key Experiments for Demonstrating Selectivity in Method Validation
| Experiment Type | Purpose | Acceptance Criteria (Example) | Applicable Techniques |
|---|---|---|---|
| Blank Matrix Analysis | To verify the absence of endogenous interference. | No significant response (e.g., < 20% of analyte response at LLOQ) at the retention time of the analyte. | Chromatography, Spectrometry |
| Interference Spiking | To check for interference from known compounds (e.g., drugs, metabolites). | Resolution between analyte and closest interfering peak > 2.0. Signal change < ±5% for accuracy. | Chromatography, LBAs |
| Forced Degradation | To demonstrate stability-indicating properties and resolution from degradants. | Peak purity of analyte confirmed; all degradants are baseline resolved. | Chromatography (primarily) |
| Cross-Reactivity | To ensure antibodies or receptors do not bind to similar molecules. | Cross-reactivity < 1% for all listed related compounds. | Ligand Binding Assays |
The Scientist's Toolkit: Essential Reagents and Materials
The experimental protocols for establishing selectivity require carefully selected reagents and materials to generate reliable and defensible data.
Table 3: Essential Research Reagent Solutions for Selectivity/Specificity Studies
| Reagent / Material | Function in Selectivity Assessment | Critical Quality Attributes |
|---|---|---|
| High-Purity Analytical Reference Standard | Serves as the benchmark for the target analyte's behavior (retention time, signal). | Certified purity (>98%), proper identity confirmation (e.g., via NMR, MS). |
| Potential Interferent Standards | Used to challenge the method's ability to distinguish the analyte from similar compounds. | Should include known impurities, degradation products, metabolites, and common co-formulants. |
| Blank Matrix | The analyte-free biological fluid or sample material used to assess background interference. | Should be representative of the test samples; for bioanalysis, use from at least 6 different sources. |
| Stressed Samples (Forced Degradation) | Generated by exposing the analyte to stress conditions to create potential degradants for interference testing. | Should typically produce 5-20% degradation; avoid excessive degradation (>30%) which may create secondary degradants. |
| Chromatographic Columns | The stationary phase for separation; critical for achieving resolution between analyte and interferents. | Multiple columns from different batches/lots should be evaluated during robustness testing. |
| Specific Antibodies (for LBAs) | The binding reagent that provides the basis for recognition and measurement in ligand binding assays. | High affinity and, crucially, low cross-reactivity against a panel of structurally similar molecules. |
Visualization of the Specificity-Selectivity Relationship
The conceptual relationship between selectivity and specificity, as defined by IUPAC, can be visualized as a spectrum or hierarchy of analytical discrimination.
Diagram 2: The Specificity-Selectivity Hierarchy
This diagram illustrates that selectivity is a scalable property. A method can have poor, moderate, or high selectivity. Specificity sits at the apex of this hierarchy as the theoretical ideal of perfect selectivity—a state where the method is affected by one and only one analyte. In practice, the goal of method development is to achieve sufficient selectivity for the intended purpose, acknowledging that absolute specificity may be an unattainable ideal for most techniques when faced with the infinite complexity of real-world samples [18].
The debate between specificity and selectivity is more than a matter of terminology; it reflects a fundamental understanding of the capabilities and limitations of analytical methods. IUPAC's stance—promoting selectivity as the preferred, scalable term and reserving specificity for the ultimate, ideal state—provides a scientifically rigorous framework. This perspective encourages a more nuanced and evidence-based approach to method validation, where scientists actively investigate and document a method's ability to distinguish an analyte from a defined panel of potential interferents, rather than simply claiming "specificity."
For the drug development professional, this means that validation protocols must be thoughtfully designed to challenge the method with all reasonably expected interferents. The experimental protocols outlined in this paper—from forced degradation studies to interference testing—provide a roadmap for this essential work. As analytical technologies continue to evolve, pushing the boundaries of sensitivity and resolution, the practical performance of our methods will increasingly approach the theoretical ideal of specificity. However, a clear understanding of the distinction, grounded in IUPAC recommendations, will remain vital for scientific accuracy, regulatory compliance, and the advancement of analytical science.
In the rigorous world of analytical science, particularly within pharmaceutical development, the terms "specificity" and "selectivity" are often used interchangeably. However, they describe distinct method characteristics whose proper identification is critical for method validation integrity. The International Council for Harmonisation (ICH) and regulatory bodies like the U.S. Food and Drug Administration (FDA) provide a framework for validation, defining fundamental performance characteristics that ensure a method is suitable for its intended purpose [21] [22]. Within this framework, understanding whether a method is specific or selective dictates the entire validation strategy, influencing experimental design, acceptance criteria, and ultimately, the degree of confidence in the generated data.
Specificity is the ability of a method to measure the analyte accurately and exclusively in the presence of other components that are expected to be present in the sample matrix. It is the highest expression of method discrimination, often described as "absolute selectivity" [22]. A specific method can unequivocally assess the analyte without interference from impurities, degradation products, or the sample matrix itself. In contrast, selectivity is the ability of the method to measure the analyte accurately in the presence of a smaller number of potential interfering substances. A selective method can distinguish the analyte from a limited set of other analytes or interferences, but may not be immune to all components in a complex matrix. This distinction is not merely semantic; it is foundational to demonstrating that an analytical procedure can generate reliable results that support critical decisions in drug development, manufacturing, and quality control [21].
Regulatory and Scientific Significance
From a regulatory perspective, the distinction between specificity and selectivity is embedded within modern analytical guidelines. The ICH Q2(R2) guideline on analytical procedure validation mandates the evaluation of specificity as a core parameter, requiring that it be demonstrated for identification tests, impurity tests, and assay methods [21] [22]. For identification, the method must be able to discriminate between compounds of closely related structure. For purity and assay methods, it must demonstrate a lack of interference from other components.
The adoption of a lifecycle approach to method validation, as emphasized in the modernized ICH Q2(R2) and ICH Q14 guidelines, further elevates the importance of this distinction [21]. Under this model, validation is not a one-time event but a continuous process beginning with method development. Defining a method's discriminatory power—as either specific or selective—at the Analytical Target Profile (ATP) stage ensures that the subsequent validation plan is scientifically sound and risk-based. A method intended for the release of a final drug product, where the sample matrix is well-defined but complete, requires a demonstration of specificity. A method used for in-process testing or for a biomarker in a complex biological matrix may be validated as selective, with a clear understanding of its limitations [23]. Mischaracterization at this stage can lead to a validation package that fails to adequately challenge the method, creating regulatory and product quality risks.
Table 1: Key Differences Between Specificity and Selectivity
| Feature | Specificity | Selectivity |
|---|---|---|
| Core Definition | Measures only the target analyte with no interference from other components. | Measures the target analyte in the presence of a limited number of potential interferences. |
| Scope | The highest degree of selectivity; "absolute" [22]. | A relative measure of discrimination; exists on a spectrum. |
| Interferences Considered | All components expected to be present (e.g., impurities, degradants, matrix) [22]. | A defined set of potential interfering substances. |
| Regulatory Emphasis | Explicitly required by ICH Q2(R2) for identification, assay, and impurity tests [21] [22]. | Often discussed as a broader concept; demonstrated when full specificity is not achievable. |
| Typical Application | Finished product release testing, stability-indicating methods. | In-process controls, biomarker assays in complex matrices [23]. |
Experimental Protocols for Demonstrating Specificity and Selectivity
The experimental design for proving a method's discriminatory power depends on its intended claim and the nature of the analyte and matrix. The following protocols outline the standard methodologies cited in industry practices and regulatory guidances.
Protocol for Specificity Testing
The objective is to prove the method's response is due solely to the target analyte, even when other components are present.
Materials and Reagents:
- Analyte of Interest (Drug Substance): High-purity reference standard.
- Forced Degradation Samples: Samples of the drug substance and product stressed under conditions of light, heat, acid, base, and oxidation.
- Sample Matrix Placebo: The formulation blank containing all excipients but no active ingredient.
- Known Impurities and Synthetic Intermediates: Authentic standards, where available.
Methodology:
- Chromatographic Peak Purity Assessment: For chromatographic methods (e.g., HPLC-UVDAD, LC-MS), inject the following and compare the chromatograms:
- Analyte Standard: A known concentration of the pure analyte.
- Placebo/Blank Matrix: To confirm no interfering peaks co-elute with the analyte.
- Forced Degradation Samples: To demonstrate that the analyte peak is pure and free from overlapping peaks from degradants. Peak purity is assessed using a photodiode array detector (DAD) or mass spectrometry (MS) to confirm a homogeneous peak.
- Spiked Mixtures: The placebo or blank matrix spiked with known impurities and the analyte. This confirms baseline separation of the analyte peak from all potential interferents [22].
Quantitative Recovery (for Assays): Compare the results for the analyte in the presence and absence of the other components. The recovery of the analyte should be within validated accuracy limits (e.g., 98-102%), demonstrating that the matrix or impurities do not suppress or enhance the analyte's signal.
Detection and Quantification of Impurities: The method should be capable of detecting and quantifying known and unknown impurities at or below the reporting threshold, with clear resolution from the main analyte peak.
Protocol for Selectivity Testing
The objective is to prove the method can distinguish and quantify the analyte in the presence of a defined set of other analytes or potential interferences.
Materials and Reagents:
- Target Analyte(s): Reference standard.
- Potential Interferents: A defined list of compounds that are structurally similar, metabolically related, or known to be present in the sample type (e.g., concomitant medications, key endogenous compounds for biomarker assays).
- Representative Sample Matrix: A pool of biological fluid (e.g., plasma, serum) or other complex matrix.
Methodology:
- Resolution of Analyte Mixtures: Prepare a mixture containing the target analyte and all defined potential interferents at their expected maximum concentrations. Analyze the mixture and demonstrate that the method provides baseline resolution (resolution factor, Rs > 1.5) between the analyte peak and each interferent peak.
Interference Check in Matrix: Analyze at least six independent sources of the blank sample matrix (e.g., plasma from six different donors).
- Ensure that at the retention time of the analyte, the response from the blank matrix is less than 20% of the lower limit of quantitation (LLOQ) for the analyte.
- Ensure that no endogenous compounds co-elute with the analyte or any of the defined interferents.
Cross-Reactivity Assessment (for Ligand Binding Assays - LBAs): Test the method's response against the panel of potential interferents. A significant response (e.g., >5% of the signal at the LLOQ) indicates cross-reactivity and a limitation in the method's selectivity, which must be reported and justified for the Context of Use [23].
Table 2: Key Reagents and Their Functions in Specificity/Selectivity Testing
| Reagent / Material | Function in Validation |
|---|---|
| Reference Standard | Serves as the benchmark for the pure analyte's properties (retention time, spectral profile). |
| Placebo / Blank Matrix | Identifies interference from the sample matrix or formulation excipients. |
| Forced Degradation Samples | Challenges the method's ability to distinguish the analyte from its degradation products. |
| Authentic Impurity Standards | Used to verify resolution and confirm the method can detect and quantify known impurities. |
| Independent Matrix Lots | Assesses variability in endogenous components that could affect method selectivity. |
A Framework for Decision-Making
The following workflow diagrams the logical process for determining and validating a method's discriminatory power, integrating the concepts of risk and Context of Use.
Implications for Different Analytical Fields
The specificity/selectivity distinction has varying implications across analytical applications.
Pharmaceutical Quality Control (PK Assays)
For pharmacokinetic (PK) assays, which measure drug concentration, the analyte is a fully characterized reference standard (the drug itself). The matrix, while complex, is consistent (e.g., human plasma). The goal is to achieve specificity by demonstrating no interference from the matrix or metabolites. The ICH M10 framework provides a prescriptive path for this, often using spike-recovery experiments [23].
Biomarker Assay Validation
This area highlights the critical nature of the distinction. Biomarker assays measure endogenous molecules for which a pristine reference standard identical to the analyte may not exist [23]. The sample matrix is highly variable. Achieving absolute specificity is often impossible. Therefore, a "fit-for-purpose" approach is used, and methods are validated for selectivity [23]. The validation must demonstrate that the method can reliably measure the biomarker in the presence of known, variable interferents. Key experiments include parallelism assessment (to show the calibrator behaves like the endogenous analyte) and testing in many individual matrix lots to establish the range of selectivity [23]. The 2025 FDA BMVB guidance explicitly recognizes these differences and discourages the blind application of the ICH M10 PK framework to biomarker assays [23].
The integrity of an analytical method is inextricably linked to a scientifically rigorous and honest assessment of its discriminatory capabilities. The distinction between specificity and selectivity is not a pedantic exercise but a fundamental principle of method validation. Correctly characterizing a method forces a deeper understanding of the analyte, the matrix, and the method's technical limitations. As the regulatory landscape evolves towards a more holistic, lifecycle-based approach grounded in Science and Risk-Based Planning [21], this clarity becomes paramount. By meticulously defining and demonstrating whether a method is specific or selective, scientists provide the transparency and robust evidence that regulators demand, ensuring that analytical data is trustworthy and fit-for-purpose in the journey to deliver safe and effective medicines.
From Theory to Practice: Assessing Specificity and Selectivity in Analytical Methods
Experimental Designs for Demonstrating Specificity in Assay and Impurity Methods
In analytical method validation, the concepts of specificity and selectivity are fundamental, yet they are often used interchangeably despite having distinct meanings. Specificity refers to the ability of a method to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradants, or matrix components [1]. It is the ability to measure accurately and specifically the analyte of interest despite these potential interferents [24]. Selectivity, meanwhile, describes the ability of the method to differentiate and quantify multiple analytes in a mixture, requiring the identification of all components rather than just the target analyte [1] [11].
This distinction frames a critical challenge in pharmaceutical development: designing experimental approaches that adequately demonstrate a method's capacity to measure the intended analyte without interference from closely related substances. This technical guide explores advanced experimental designs for establishing method specificity, particularly for potency assays and impurity methods, providing researchers with structured approaches for generating defensible validation data.
Conceptual Framework: Specificity Versus Selectivity
Regulatory Definitions and Distinctions
According to ICH guidelines, specificity is the ability to assess unequivocally the analyte in the presence of components that may be expected to be present [1]. A commonly used analogy describes specificity as identifying "the correct key for the lock" among a bunch of keys, without needing to identify all other keys [1] [11]. Selectivity, while similar, requires the identification of all components in a mixture [11]. The International Union of Pure and Applied Chemistry (IUPAC) actually recommends the term "selectivity" over "specificity" in analytical chemistry, recognizing that few methods respond to only a single analyte [1].
For impurity methods, specificity requires demonstrating that the method can separate and quantify individual impurities from each other and from the main analyte, often through resolution measurements between closely eluting peaks [24]. For assay methods, specificity must demonstrate that the measured response is due solely to the target analyte, achieved through studies showing no interference from blank matrices, placebos, impurities, or degradation products [25] [24].
Analytical Significance in Pharmaceutical Development
The demonstration of specificity is crucial across multiple application contexts in drug development. For identification tests, specificity is absolutely necessary to ensure only the target analyte is detected without cross-reactions [1]. For assay and impurity tests, specificity ensures accurate quantification of the active ingredient and reliable detection of impurities at low levels [24]. In bioassays, specificity confirms that the measured signal genuinely reflects the biological activity of the molecule, without interference from formulation buffers, media, or degraded product [25].
Failure to adequately demonstrate specificity can lead to inaccurate potency assignments, failure to detect critical impurities, and ultimately, regulatory objections to method implementation. The following sections detail experimental designs to rigorously address these challenges.
Experimental Designs for Specificity Demonstration
Chromatographic Method Specificity Protocols
For chromatographic methods, specificity is typically demonstrated through resolution measurements between the analyte peak and potential interferents. The experimental protocol involves analyzing several sample solutions [24]:
Placebo/Blank Analysis: The sample matrix without the analyte demonstrates no interfering peaks at the retention time of the target analyte.
Forced Degradation Studies: The drug substance or product is subjected to stress conditions (acid, base, oxidation, thermal, photolytic) to generate degradants, followed by demonstration of separation between the analyte and degradation peaks.
Spiked Mixtures: The analyte is spiked with available impurities, excipients, or related compounds to demonstrate resolution from all potential interferents.
Peak Purity Assessment: Using photodiode array (PDA) or mass spectrometry (MS) detection to demonstrate peak homogeneity and absence of coeluting substances [24].
A key acceptance criterion for specificity is the resolution of the two most closely eluted compounds, typically requiring a resolution factor (Rs) greater than 1.5 between the analyte and nearest eluting potential interferent [24].
Bioassay Specificity and Selectivity Approaches
For bioassays, specificity evaluation follows different principles centered on biological response rather than physical separation. Two primary approaches are used [25]:
Signal Specificity: Demonstration that only the specific protein generates the measured signal, while blanks, placebos, or other proteins generate no response.
Interference Testing: Evaluation of potential interference from materials such as media, formulation buffers, or forced degradation materials by spiking these into the assay system and observing any shift in the response.
In cell-based bioassays, specificity is further supported by demonstrating that the measured response aligns with the known biological mechanism of action, providing additional confidence that the signal reflects the intended activity [25].
Advanced Computational and DoE Approaches
Recent advances incorporate computational modeling and Design of Experiments (DoE) for more robust specificity demonstrations. Biophysical models trained on high-throughput selection data can disentangle different binding modes, enabling the design of antibodies with customized specificity profiles [26]. This approach allows discrimination between even structurally and chemically similar ligands.
DoE approaches systematically evaluate multiple assay parameters simultaneously to establish method robustness and identify critical factors affecting specificity [27]. A well-executed DoE study can efficiently characterize the design space where the method maintains specificity despite normal operational variations [25].
Figure 1: Experimental Workflow for Specificity Demonstration
Quantitative Assessment and Acceptance Criteria
Specificity Acceptance Criteria for Different Method Types
The demonstration of specificity requires defined acceptance criteria that vary based on the method type and its intended use. The following table summarizes key criteria across different analytical contexts:
Table 1: Specificity Acceptance Criteria for Different Method Types
| Method Type | Specificity Evidence | Acceptance Criteria | Regulatory Reference |
|---|---|---|---|
| Identification | Ability to discriminate between compounds of similar structure | Comparison to known reference material; no false positives/negatives | ICH Q2(R1) [1] |
| Assay | Resolution from closely eluting impurities | Resolution factor (Rs) ≥ 1.5 between analyte and nearest impurity | USP <621> [24] |
| Impurity Test | Separation and individual quantification of all specified impurities | Baseline separation (Rs ≥ 1.5) between all impurity pairs | ICH Q3B [24] |
| Bioassay | Signal generated only by active ingredient; no matrix interference | ≤ 10% change in accuracy in presence of interferents | USP <1033> [25] |
Statistical Parameters for Specificity Demonstration
The quantitative assessment of specificity incorporates statistical measures to establish method reliability:
Table 2: Statistical Measures for Specificity Assessment
| Parameter | Calculation | Target Value | Application Context |
|---|---|---|---|
| Resolution (Rs) | Rs = 2(t₂ - t₁)/(w₁ + w₂) | ≥ 1.5 | Chromatographic separations |
| Peak Purity | Spectral similarity index or purity angle | Purity angle < purity threshold | PDA or MS detection |
| Signal Interference | % Interference = (Responsewithinterferent - Responsealone)/Responsealone × 100 | ≤ 10% | Bioassays, matrix effects |
| Recovery | % Recovery = (Measured concentration/Spiked concentration) × 100 | 90-110% | Specificity in complex matrices |
For chromatographic methods, specificity is typically demonstrated by injecting samples containing the analyte spiked with potential interferents (impurities, excipients, degradation products) and showing that the resolution between the analyte peak and the closest eluting potential interferent is greater than 1.5 [24]. For bioassays, specificity may be demonstrated by showing minimal change in accuracy (typically ≤ 10%) when potential interferents are present in the sample matrix [25].
Case Study: Specificity in Bioassay Development
Experimental Design for Cell-Based Bioassay Specificity
A comprehensive approach to bioassay specificity was demonstrated in a qualification study for a cell-based bioassay measuring cytotoxic activity of an antibody-drug conjugate [27]. The experimental design incorporated:
- Multiple sample preparations at different potency levels (50%, 71%, 100%, 141%, 200%) to evaluate specificity across the method range
- Fractional factorial design examining five critical assay parameters (cell density and four timing parameters) at low, middle, and high levels
- Statistical analysis using a random-effects model to estimate variability components from different analysts, days, preparations, and plates
The study evaluated specificity through interference testing by examining whether critical assay parameters significantly affected the relative potency results. The lack of statistically significant main effects or interaction terms in the statistical model for relative potency (p-values ranging from 0.12) demonstrated assay robustness and specificity across the examined operational ranges [27].
Computational Approaches for Antibody Specificity Design
Advanced computational methods now enable the design of antibodies with customized specificity profiles. One approach involves:
- Phage display experiments with selection against various combinations of closely related ligands
- High-throughput sequencing to monitor antibody library composition at each selection step
- Biophysical modeling to identify distinct binding modes associated with specific ligands
- Computational generation of antibody variants not present in the initial library with defined specificity profiles
This approach successfully addressed one of the most challenging tasks in the field: designing antibodies capable of discriminating between structurally and chemically similar ligands [26]. The model successfully disentangled different binding modes even when associated with chemically very similar ligands, enabling computational design of antibodies with either specific high affinity for a particular target or cross-specificity for multiple targets.
Figure 2: Computational Workflow for Antibody Specificity Design
Essential Research Reagents and Materials
Key Reagent Solutions for Specificity Studies
Successful specificity demonstration requires carefully selected reagents and materials designed to challenge the method with potential interferents:
Table 3: Essential Research Reagents for Specificity Evaluation
| Reagent/Material | Function in Specificity Assessment | Application Context |
|---|---|---|
| Placebo Formulation | Contains all excipients without active ingredient to demonstrate no matrix interference | Assay methods, impurity methods |
| Forced Degradation Samples | Stressed samples containing degradation products to demonstrate separation from main analyte | Stability-indicating methods |
| Available Impurities/Related Compounds | chemically synthesized impurities for spiking studies to demonstrate resolution | Impurity methods, assay methods |
| Alternative Protein/Enzyme Preparations | Structurally similar proteins to demonstrate specificity of biological response | Bioassays, binding assays |
| Matrix Components | Serum, plasma, or tissue extracts to evaluate matrix effects in biological samples | Bioanalytical methods |
| Cross-Reactive Analytes | Structurally similar compounds likely to cross-react to demonstrate discrimination | Immunoassays, receptor binding assays |
These reagents enable the systematic challenge of the analytical method to demonstrate that the measured response is specific to the target analyte despite the presence of structurally similar compounds, matrix components, or degradation products [25] [24].
The demonstration of specificity requires carefully designed experiments that challenge the method with potential interferents relevant to the sample matrix and analytical context. For chromatographic methods, this typically involves forced degradation studies and resolution measurements between closely eluting peaks. For bioassays, specificity is demonstrated through interference studies and biological relevance. Advanced approaches incorporating computational modeling and DoE provide more robust specificity demonstrations, enabling methods that maintain performance characteristics despite normal operational variations. Properly designed specificity studies generate defensible data that establishes method reliability for its intended use throughout the method lifecycle.
Techniques for Establishing Selectivity in Complex Biological Matrices
In analytical method validation, the terms "specificity" and "selectivity" are often used interchangeably, but they represent distinct concepts crucial for assays in complex biological matrices. Specificity refers to the ability of a method to measure unequivocally a single analyte in the presence of other components expected to be present in the sample matrix. It describes the degree of interference by other substances while analyzing the target analyte. A specific method identifies the correct "key" among a bunch of other keys without needing to identify the other keys [1].
Selectivity, while related, is a broader concept. It describes the degree to which a method can quantify an analyte in the presence of other target analytes or matrix interferences. For a method to be selective, the identification of all relevant components in a mixture is essential. It is the parameter that ensures a method can accurately measure multiple analytes simultaneously without cross-reactivity or interference [1] [12]. In the context of complex biological samples like serum, plasma, or tissue homogenates—which contain various proteases, inhibitors, and other interfering substances—achieving high selectivity becomes a significant analytical challenge [28].
Core Challenges in Complex Matrices
Biological matrices such as human serum present substantial analytical challenges due to their complex composition. Serum contains a diverse array of proteases, inhibitors, and other biomolecules that regulate physiological processes and metabolism [28]. When developing biosensors or assays based on specific reactions, such as proteolytic cleavage, these endogenous components can severely interfere with the target analyte's activity, leading to two main problems:
- Reduced Selectivity: Multiple enzymes or components may recognize and react with the same substrate or detection reagent. For instance, multiple Matrix Metalloproteinases (MMPs), such as MMP-1, -7, -8, and -9, can recognize and cleave identical amino acid sequences, making it difficult to attribute a signal to one specific protease [28].
- Loss of Detection Capability: The presence of endogenous inhibitors can bind to or suppress the activity of the target analyte, resulting in false negatives or a significant underestimation of the analyte's concentration [28].
Overcoming these limitations often requires sophisticated sample handling or integrated assay designs that isolate the analyte and minimize matrix effects.
Technical Approaches for Enhancing Selectivity
Several techniques can be employed to achieve the high selectivity required for accurate analysis in complex biological matrices.
Affinity Capture and Isolation
A highly effective approach involves the affinity capture of the target analyte using surface-immobilized antibodies or other capture agents, followed by washing steps. This strategy physically isolates the target from the complex sample matrix before detection [28].
- Procedure: The sample is incubated with a solid support (e.g., an electrode, magnetic bead, or well plate) coated with a capture antibody specific to the target protein. After incubation, unbound components are removed through washing. The purified analyte is then detected.
- Advantages: This method selectively enriches the target and effectively minimizes interference from other proteases, inhibitors, and matrix components that are removed during washing [28]. It is particularly advantageous for electrochemical detection, as the localized generation of reaction products near the electrode surface can enhance the analytical signal [28].
Orthogonal Separation Techniques
Utilizing separation mechanisms that are orthogonal (i.e., based on different physicochemical principles) to standard methods can significantly enhance selectivity.
- Capillary Electrophoresis-Mass Spectrometry (CE-MS): CE separates molecules based on their charge and size, which is orthogonal to the hydrophobicity-based separation of liquid chromatography (LC). When coupled with MS, this provides unique selectivity, especially for charged and polar molecules. This is highly valuable for analyzing hydrophilic metabolites in metabolomics or post-translationally modified peptides in proteomics, which are challenging for reversed-phase LC [29].
- Nanobore Chromatography: The growth of quantitative approaches employing nanobore LC-MS stimulates the need for robust separations. Selecting the correct stationary phase (e.g., C18, C8, PFP, CN) is a key part of method development for improving selectivity, as predicting the selectivity of a specific phase for a specific analyte is virtually impossible and must be determined empirically [30].
Cascade Amplification Systems
Cascade reactions can be designed to enhance both sensitivity and selectivity. These are multi-step reactions where the initial recognition and activation by the target analyte triggers a subsequent, highly amplified detection signal.
- Enzyme Activation Cascade: An innovative approach uses an auto-inhibited enzyme, such as a bioengineered β-lactamase connected to its inhibitor protein via a peptide linker. The enzyme is inactive in its zymogen form. Proteolytic cleavage of the peptide linker by a specific target protease (e.g., MMP-2) physically separates the inhibitor from the enzyme, restoring its catalytic activity. The activated enzyme then rapidly generates a detectable product from its substrate, amplifying the initial recognition event [28].
- Benefit: This system can improve selectivity by linking detection to two specific events (protease cleavage and enzyme activation) and enhances sensitivity through signal amplification [28].
Detailed Experimental Protocol: A Case Study
The following workflow, which combines affinity capture with a cascade reaction for the selective detection of Matrix Metalloproteinase-2 (MMP-2) in human serum, exemplifies the application of these principles [28].
The diagram below illustrates the sequential steps involved in this selective detection method.
Materials and Reagents
Table 1: Key Research Reagent Solutions for Selective MMP-2 Detection
| Reagent/Material | Function/Description | Source/Example |
|---|---|---|
| Anti-MMP-2 IgG | Capture antibody for specific affinity isolation of MMP-2 from the sample matrix. | R&D Systems, Inc. (e.g., AF902) [28] |
| Auto-inhibited β-lactamase | Engineered zymogen; the reporter enzyme. Inactive until cleaved by MMP-2. | Bioengineered construct with β-lactamase and BLIP connected by an MMP-2 cleavable linker [28] |
| Nitrocefin | Chromogenic/electroactive substrate for β-lactamase. Conversion generates a detectable signal. | Sigma-Aldrich [28] |
| Human Serum | Complex biological matrix for the assay, containing interfering substances. | Commercial source (e.g., Sigma-Aldrich) [28] |
| Assay Buffer (Tris, Brij-35, NaCl, CaCl₂) | Provides optimal pH, ionic strength, and conditions for maintaining MMP-2 activity and reducing non-specific binding. | Standard chemical suppliers [28] |
| Bovine Serum Albumin (BSA) | Used as a blocking agent to minimize non-specific adsorption to surfaces. | Sigma-Aldrich [28] |
Step-by-Step Procedure
Affinity Capture:
- Incubate the human serum sample with an electrode modified with anti-MMP-2 capture antibodies.
- Wash the electrode thoroughly with an appropriate buffer (e.g., Tris buffer containing Brij-35) to remove unbound proteins, inhibitors, and other interfering substances present in the serum [28].
Cascade Reaction Initiation:
- Incubate the electrode with a solution containing the engineered auto-inhibited β-lactamase.
- The captured MMP-2 specifically cleaves the peptide linker on the β-lactamase zymogen, releasing the inhibitor (BLIP) and activating the β-lactamase enzyme.
Signal Generation and Detection:
- Add the β-lactamase substrate (e.g., nitrocefin). The activated enzyme rapidly converts the substrate into an electroactive product (open-nitrocefin).
- Perform an electrochemical measurement (e.g., amperometry or voltammetry) to quantify the generated electroactive product. The signal intensity is proportional to the MMP-2 activity captured from the sample.
Selectivity Assessment Protocol
To validate the selectivity of the method, the following tests should be performed:
- Interference Test: Analyze samples spiked with structurally similar or functionally related enzymes (e.g., MMP-7, MMP-8, MMP-9, trypsin, plasmin) at physiologically relevant concentrations. A selective method should show minimal response to these non-target analytes [28].
- Matrix Effect Test: Analyze the target analyte (MMP-2) in the presence of the biological matrix (serum) and compare the response to that in a clean buffer. Calculate the percentage recovery to demonstrate that the matrix does not suppress or enhance the signal [12].
- Blank Sample Analysis: Use reagent blanks (containing all reagents but no sample) and matrix blanks (containing the sample matrix without the analyte) to assess the background signal and confirm the measured signal originates from the analyte [12].
Data Analysis and Performance Metrics
The performance of a selective method is quantified using specific validation parameters. The data from the MMP-2 case study can be summarized as follows:
Table 2: Key Analytical Performance Metrics for Selective MMP-2 Detection
| Performance Metric | Result / Value | Experimental Detail |
|---|---|---|
| Limit of Detection (LOD) | Successfully determined in human serum | Defined as the lowest concentration of analyte that can be reliably detected (Signal/Noise ≈ 3) [28] [12]. |
| Selectivity vs. other MMPs | Enhanced selectivity achieved against MMP-7, -8, and -9 | Demonstrated via affinity capture, which isolated MMP-2 and reduced cross-reactivity [28]. |
| Recovery in Serum | Effectively minimized interference from serum inhibitors | Assessed by comparing the signal in serum vs. buffer, showing the method's robustness to matrix effects [28]. |
| Linearity and Range | Demonstrated across the analytical procedure | A linear relationship between electrochemical signal and MMP-2 concentration was established, typically using a minimum of five concentration points [12]. |
Establishing selectivity in complex biological matrices is a multi-faceted challenge that requires strategic method design. As demonstrated, techniques such as affinity capture, orthogonal separations, and cascade reaction systems are powerful tools for isolating the target analyte and mitigating interference. The case study on MMP-2 detection underscores that a combination of these techniques—rather than relying on a single approach—can yield highly selective and sensitive assays. Validating this selectivity through rigorous interference and matrix effect testing is paramount for generating reliable data in research and drug development.
The Role of Forced Degradation Studies in Specificity Assessment
In the framework of analytical method validation, specificity is the ability of a method to measure the analyte accurately and specifically in the presence of other components that may be expected to be present in the sample matrix, such as impurities, degradants, or excipients [31] [32]. Forced degradation studies, also known as stress testing, serve as a critical practical tool to demonstrate this parameter. These studies involve the deliberate and exaggerated degradation of a drug substance or product under a variety of stress conditions to generate samples containing potential degradants [33] [34]. The core objective is to challenge the analytical method by proving its capability to separate and quantify the active pharmaceutical ingredient without interference from its degradation products, thus confirming its stability-indicating nature [35] [34]. This article explores the integral role of forced degradation studies in assessing the specificity of analytical methods, a cornerstone for ensuring drug product quality, safety, and efficacy.
The Scientific and Regulatory Foundation of Forced Degradation
Objectives and Strategic Importance
Forced degradation studies are a regulatory expectation and a scientific necessity during drug development [33]. Their primary objectives in the context of specificity assessment include:
- To establish degradation pathways and identify degradation products: By understanding how a molecule degrades, scientists can anticipate potential impurities and develop methods to detect them [33] [36].
- To validate the stability-indicating nature of analytical methods: This is the foremost goal for specificity assessment. The method must demonstrate that it can accurately measure the analyte of interest amid all other degradation-induced components [35] [34].
- To elucidate the structure of degradation products: Identifying major degradants helps in understanding their potential toxicological impact [33] [35].
- To determine the intrinsic stability of the molecule: This knowledge informs formulation development, packaging, and storage condition selection [33] [37].
From a regulatory standpoint, guidelines from the International Council for Harmonisation (ICH) indicate that manufacturers should propose stability-indicating methodologies that can detect changes in the identity, purity, and potency of the product [34]. While ICH Q1A(R2) suggests stressing products under conditions like hydrolysis, oxidation, photostability, and temperature, the guidelines are purposefully general, allowing for a science-based approach tailored to the specific product [35] [36].
Specificity within the Method Validation Framework
Specificity is a fundamental validation parameter that must be established before a method is deployed for stability studies or release testing [31] [32]. A method lacking specificity can lead to inaccurate potency results or a failure to detect critical impurities, compromising patient safety and product quality. Forced degradation studies provide the most rigorous challenge for demonstrating specificity by generating real-world samples containing the very impurities the method is designed to monitor throughout the product's shelf life [34].
Designing Forced Degradation Studies for Specificity Assessment
A well-designed forced degradation study is paramount for a meaningful specificity assessment. The strategy involves selecting appropriate stress conditions, achieving a sufficient level of degradation, and using representative materials.
Critical Stress Conditions and Methodologies
A minimal list of stress factors should be investigated to cover major degradation pathways. The conditions should be more severe than those used in accelerated stability studies but should aim to avoid secondary degradation that would not be relevant under normal storage conditions [33] [34]. The following table summarizes common forced degradation conditions and their implementation.
Table 1: Standard Forced Degradation Conditions and Protocols
| Stress Condition | Typical Experimental Parameters | Target Degradation Pathways |
|---|---|---|
| Acid Hydrolysis | 0.1 - 1.0 M HCl at 40-80°C for several hours to days [35] [38] | Peptide bond cleavage (fragmentation), especially at Asp-Pro and Asp-Gly bonds [36] |
| Base Hydrolysis | 0.1 - 1.0 M NaOH at 40-80°C for several hours to days [35] [38] | Deamidation (Asn, Gln), hydrolysis, and racemization [36] |
| Oxidation | 3-30% Hydrogen Peroxide (H₂O₂) at room or elevated temperature [33] [35] | Oxidation of Met, Cys, His, Trp, and Tyr side chains [36] |
| Thermal Stress | 40-80°C in dry or humidified states (e.g., 75% Relative Humidity) for up to 14 days [39] [33] | Aggregation (covalent and non-covalent), deamidation, hydrolysis [39] |
| Photolysis | Exposure to UV (320-400 nm) and visible light per ICH Q1B guidelines [33] [35] | Free radical-mediated oxidation, aggregation, and backbone cleavage [36] |
Optimizing the Degradation Extent and Sample Selection
A key consideration is determining the optimal level of degradation. A degradation of 5-20% is generally considered adequate for validating chromatographic assays [33] [34]. Under-stressing may not generate sufficient degradants to challenge the method, while over-stressing can produce secondary degradation products not representative of real-world stability profiles [33] [36].
Forced degradation studies should be performed on a single batch of drug substance or drug product that is representative of the final manufacturing process [35] [34]. It is considered a one-time study and is not part of the formal stability protocol. Including relevant controls, such as stressed placebo and unstressed drug product, is essential to distinguish degradation products of the active ingredient from those that may arise from excipients [34].
Analytical Techniques and Data Interpretation for Specificity
The Analytical Toolkit
Due to the complexity of potential degradation pathways, especially for biologics, a combination of orthogonal analytical techniques is required to fully assess specificity and characterize degradants [39] [36]. The following table outlines key techniques and their specific roles in evaluating degradation.
Table 2: Key Analytical Techniques for Assessing Specificity in Forced Degradation Studies
| Analytical Technique | Primary Role in Specificity Assessment | Degradation Products Detected |
|---|---|---|
| Size-Exclusion Chromatography (SE-HPLC/UPLC) | Separates and quantifies monomeric protein from soluble aggregates and fragments [39] [34] | High-molecular-weight (HMW) aggregates, low-molecular-weight (LMW) fragments [39] |
| Reversed-Phase Chromatography (RP-HPLC/UPLC) | Assesses purity and separates variants based on hydrophobicity [38] [36] | Oxidized species, clipped variants, other product-related impurities [36] |
| Capillary Electrophoresis (CE-SDS) | Provides purity and impurity analysis under denaturing conditions [39] | Protein fragments and aggregates [39] |
| Ion-Exchange Chromatography (IEX) / imaged Capillary IEF (icIEF) | Separates charge variants of the protein [39] | Deamidated, acetylated, or sialylated forms; charge heterogeneity [39] |
| Peptide Mapping | Provides detailed characterization of chemical modifications at the amino acid level [36] | Site-specific oxidation, deamidation, glycation [36] |
Key Data Interpretation Parameters
Interpreting data from forced degradation studies involves several critical assessments to confirm method specificity [35]:
- Peak Purity Analysis: This is a direct measure of specificity. Using diode array or mass spectrometric detectors, the purity of the analyte peak is assessed. A peak purity index of >0.995 confirms the absence of co-eluting impurities or degradants, proving the method's specificity for the analyte [35].
- Mass Balance: This involves calculating the total amount of analyte and impurities found in the stressed sample and comparing it to the amount declared. A mass balance of 90-110% provides confidence that all major degradants are being detected by the analytical method. A significant shortfall may indicate the presence of undetected degradants (e.g., non-UV absorbing aggregates), challenging the method's specificity [35].
- Resolution: The chromatographic method should demonstrate baseline resolution between the main peak and all critical degradant peaks. This ensures accurate quantification of each species without interference.
The workflow below illustrates the logical process of using forced degradation to assess method specificity.
Diagram: Specificity Assessment Workflow. This diagram outlines the process of using forced degradation studies to challenge an analytical method, leading to either confirmation or necessary optimization of its specificity.
Advanced Applications and Industry Perspectives
Use in Biopharmaceutical Comparability Studies
Forced degradation studies have a vital role beyond initial method validation, particularly in assessing comparability for biopharmaceuticals. When a change is made to the manufacturing process of a biologic, ICH Q5E recommends using forced degradation to compare the degradation profiles of the pre-change and post-change material [40]. A similar degradation profile under stress provides a higher level of assurance that the change did not adversely impact the product's quality attributes and that the validated analytical methods remain specific and applicable for the post-change product [40].
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key research reagent solutions and materials essential for conducting robust forced degradation studies.
Table 3: Essential Research Reagent Solutions for Forced Degradation Studies
| Reagent / Material | Function in Forced Degradation |
|---|---|
| Hydrochloric Acid (HCl) | Used in acid hydrolysis studies to simulate acid-catalyzed degradation, typically at 0.1 - 1.0 M concentrations [35]. |
| Sodium Hydroxide (NaOH) | Used in base hydrolysis studies to simulate base-catalyzed degradation, typically at 0.1 - 1.0 M concentrations [35]. |
| Hydrogen Peroxide (H₂O₂) | The most common oxidizing agent used to induce oxidative degradation, typically at 3-30% concentrations [33] [38]. |
| Thermostatically-Controlled Ovens/Incubators | Provide controlled thermal stress conditions at elevated temperatures (e.g., 40°C, 50°C, 80°C) for extended periods [39]. |
| ICH Q1B-Compliant Light Cabinets | Provide controlled exposure to UV and visible light to study photostability, ensuring compliance with regulatory guidance [33] [35]. |
| High-Purity Solvents & Buffers | Used for sample preparation, mobile phases, and stress condition matrices to avoid interference and unintended reactions [38]. |
Forced degradation studies are an indispensable component of modern pharmaceutical analysis, serving as the definitive experiment for demonstrating the specificity of stability-indicating methods. By strategically employing a range of stress conditions and leveraging orthogonal analytical techniques, scientists can thoroughly challenge their methods to ensure they remain accurate, reliable, and unambiguous in the presence of degradation products. As the industry advances with the adoption of Analytical Quality by Design (AQbD) and more complex modalities, the principles of well-designed forced degradation will continue to underpin the development of specific, validated methods, ultimately safeguarding public health by ensuring the quality of pharmaceutical products throughout their lifecycle.
Utilizing Blank and Spiked Samples to Evaluate Matrix Interferences
In analytical method validation, the concepts of specificity and selectivity are fundamental to demonstrating that a method is fit for its purpose. While the terms are often used interchangeably, a crucial distinction exists. Specificity refers to the ability of a method to assess the analyte unequivocally in the presence of components that may be expected to be present, such as impurities, degradants, or matrix components. It is often considered the ideal state where the method responds to one single analyte and is unaffected by other substances [1]. Selectivity, on the other hand, describes the method's ability to measure and differentiate several analytes in a mixture from other components [1] [41]. The ICH Q2(R2) guideline clarifies that "selectivity could be demonstrated when the analytical procedure is not specific," implying that a specific method is inherently selective, but a selective method may not be absolutely specific [41].
Matrix interferences represent a critical challenge to both specificity and selectivity. A matrix effect is defined as the combined effect of all components of the sample other than the analyte on the measurement of the quantity [42]. When the specific component causing a bias can be identified, it is referred to as a matrix interference [42]. These effects can manifest as signal suppression or enhancement, leading to inaccurate quantification of the target analyte and directly compromising the method's accuracy, precision, and reliability [42]. Within a thesis on specificity and selectivity, the evaluation of matrix interferences is a practical demonstration of a method's selectivity—its ability to produce accurate results for the analyte(s) of interest despite the complex sample environment. This guide provides an in-depth technical protocol for utilizing blank and spiked samples to systematically identify, quantify, and control these matrix effects.
Theoretical Foundation: The Role of Blank and Spiked Samples
Blank and spiked samples are the primary tools for diagnosing and quantifying matrix effects. They function as controlled experiments within the analytical process, allowing scientists to isolate the impact of the sample matrix from other sources of error.
Blank Samples: These are samples that contain all components of the matrix except for the target analyte. The primary blank samples used in environmental and pharmaceutical analysis include:
- Laboratory Reagent Blank (LRB) or Method Blank (MB): A clean matrix (e.g., deionized water, pure solvent) carried through the entire analytical procedure [42]. It is used to detect contamination from reagents, glassware, or the laboratory environment.
- Matrix Blank: A sample taken from the actual sample source (e.g., patient plasma, river water, formulated drug product placebo) that is confirmed to be free of the target analyte and processed through the method. It is essential for identifying signals from endogenous components that might co-elute or interfere with the analyte.
Spiked Samples: These are samples where a known quantity of the target analyte is added to either a blank matrix or the sample matrix itself. They are used to measure recovery and thus quantify matrix effects.
- Laboratory Control Sample (LCS) or Laboratory Fortified Blank (LFB): A known amount of analyte added to a clean matrix [42]. This measures the method's performance in an ideal, interference-free environment and serves as the baseline for recovery.
- Matrix Spike (MS) and Matrix Spike Duplicate (MSD): A known amount of analyte added to a sample matrix (or a separate portion of the same sample) [42]. The recovery of the spike in the MS/MSD is compared to the recovery in the LCS to determine the matrix effect.
The following workflow diagram illustrates the logical relationship between these samples and the parameters they help evaluate in an analytical method validation study.
Quantitative Evaluation of Matrix Effects
The data obtained from blank and spiked samples must be translated into quantitative metrics to objectively assess matrix effects. The following calculations are standard in the field.
Key Calculations
Percent Recovery (%R): This measures the accuracy of the measurement for the spiked analyte.
% Recovery = (Measured Concentration of Spike / Known Concentration of Spike) × 100Matrix Effect (ME%): This directly quantifies the extent of signal suppression or enhancement caused by the matrix, as defined by Matuszewski et al. [42].
ME (%) = (Matrix Spike Recovery / Laboratory Control Sample Recovery) × 100- ME = 100%: No matrix effect.
- ME > 100%: Signal enhancement.
- ME < 100%: Signal suppression.
Precision from Matrix Spike Duplicates: The relative percent difference (RPD) between the MS and MSD indicates the precision of the method in the specific sample matrix.
RPD = |(MS - MSD)| / ((MS + MSD)/2) × 100
Acceptance Criteria and Data Interpretation
Acceptance criteria for recovery and precision are often defined by regulatory methods or internal quality control procedures. The table below summarizes typical acceptance limits and the interpretation of results, providing a clear framework for evaluation.
Table 1: Interpretation of Quantitative Data from Spiked Samples
| Parameter | Calculation | Acceptance Criteria (Example) | Interpretation of Out-of-Specification Result | |||
|---|---|---|---|---|---|---|
| Laboratory Control Sample (LCS) Recovery | (Measured LCS Conc. / Known LCS Conc.) x 100 | 70-120% (Method dependent) | Indicates a fundamental problem with the method's accuracy in a clean matrix. | |||
| Matrix Spike (MS) Recovery | (Measured MS Conc. / Known MS Conc.) x 100 | 70-120% (Method dependent) | Suggests the sample matrix is affecting accuracy (bias). | |||
| Matrix Effect (ME%) | (MS Recovery / LCS Recovery) x 100 | 85-115% | ME < 100%: Signal suppression. ME > 100%: Signal enhancement. | |||
| Relative Percent Difference (RPD) | ( | MS - MSD | / Mean(MS, MSD)) x 100 | ≤ 20% (Method dependent) | Poor precision in the specific sample matrix. |
The data from these calculations feeds directly into the assessment of a method's selectivity. A method that demonstrates minimal matrix effect (ME% close to 100%) and consistent, acceptable spike recoveries across different sample matrices provides strong evidence of its selectivity. If a method can do this while also proving no interferences co-elute with the analyte (via blank analysis), it also demonstrates a high degree of specificity [1] [41].
Detailed Experimental Protocols
A robust assessment of matrix interferences requires a carefully designed experimental protocol. The following section details the methodologies for two key experiments.
Protocol 1: Initial Screening for Matrix Interferences
Objective: To identify the presence and general magnitude of matrix effects across different sample sources. Materials: See Section 6 for the Scientist's Toolkit. Procedure:
- Select a representative set of sample matrices (e.g., plasma from different donors, various river water sources, different drug product placebo formulations).
- For each matrix type, prepare the following in replicate (n=3-5):
- Matrix Blank (MB): The matrix without the target analyte.
- Matrix Spike (MS): The matrix spiked with the target analyte at a mid-level concentration within the calibration range (e.g., near the quantification level).
- Laboratory Control Sample (LCS): A clean matrix spiked at the same concentration as the MS.
- Process all samples through the entire analytical procedure, including sample preparation, chromatography, and detection.
- Analyze the data:
- Chromatograms of MBs: Inspect for any peaks that co-elute with the analyte, indicating a direct interference.
- Calculate the % Recovery for all LCS and MS samples.
- Calculate the Matrix Effect (ME%) for each matrix type.
Protocol 2: Comprehensive Matrix Effect and Recovery Assessment
Objective: To fully characterize the matrix effect and recovery across the analytical range and establish the method's selectivity. Materials: See Section 6 for the Scientist's Toolkit. Procedure:
- Select at least six different lots or sources of the sample matrix. For bioanalytical methods, this typically means individual lots of human plasma [42].
- For each matrix lot, prepare samples at two analyte concentrations (low and high, representing the lower and upper ends of the quantification range).
- For each concentration in each matrix lot, prepare the following:
- Unfortified Matrix Blank: To confirm the absence of the analyte.
- Post-Extraction Spiked Sample: The matrix blank is taken through the entire sample preparation process. After extraction, a known amount of analyte is added to the cleaned extract. This measures the matrix effect (ion suppression/enhancement in MS) without the influence of extraction recovery.
- Pre-Extraction Spiked Sample (Matrix Spike): The analyte is added to the matrix before the extraction process begins and is carried through the entire procedure. This measures the overall process efficiency (a combination of extraction recovery and matrix effect).
- Analyze all samples and calculate the matrix effect (ME%) and process efficiency (PE%) using the peak areas (PA):
ME% = (PA Post-Extraction Spike / PA of Neat Standard Solution) × 100PE% = (PA Pre-Extraction Spike / PA of Neat Standard Solution) × 100Recovery % = (PE% / ME%) × 100(if ME% and PE% are known)
The following workflow visualizes this comprehensive experimental design.
Case Study: Matrix Effects in Environmental Water Analysis by EPA Method 625
A study examining six years of quality control data for EPA Method 625 (semivolatiles) provides a compelling real-world example [42]. The researchers used an F-test to compare the standard deviations of LCS and MS/MSD recoveries to gauge the prevalence of statistically significant matrix effects.
Table 2: Example Data for Benzo[a]pyrene from EPA Method 625 [42]
| Analyte | Method | Mean LCS Recovery (%) | Mean MS/MSD Recovery (%) | Standard Deviation (LCS) | Standard Deviation (MS/MSD) | Matrix Effect (ME%) | Statistical Significance (F-test) |
|---|---|---|---|---|---|---|---|
| Benzo[a]pyrene | EPA 625 | 95.2 | 89.5 | 5.1 | 8.5 | 94.0% (Suppression) | Significant |
Findings and Interpretation: The data for benzo[a]pyrene showed a small but statistically significant matrix effect, evidenced by the larger standard deviation in the MS/MSD recoveries compared to the LCS and an ME% of 94%, indicating slight signal suppression [42]. This finding underscores that even well-established regulatory methods are not immune to matrix effects. For regulatory reporting under Method 625, if a matrix spike recovery falls outside the control limits, the associated sample results are considered "suspect" and may not be reportable for compliance [42]. This case highlights the critical importance of conducting matrix effect studies during method validation to understand the limitations and ensure the selectivity of the analytical method for its intended samples.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key materials and reagents required for conducting the experiments described in this guide.
Table 3: Essential Reagents and Materials for Matrix Interference Studies
| Item | Function / Purpose | Technical Considerations |
|---|---|---|
| Analyte Reference Standard | To prepare known, accurate spiking solutions for LCS and MS. | Must be of high purity and well-characterized (e.g., Certificate of Analysis). |
| Certified Blank Matrix | Serves as the clean matrix for LCS/LFB and preparation of calibration standards. | Should be free of the target analyte and any known interferences. For bioanalysis, charcoal-stripped plasma is often used. |
| Representative Sample Matrices | Used to prepare Matrix Blanks and Matrix Spikes for the assessment. | Should include at least 6 different lots/sources to assess variability [42]. |
| Internal Standard (IS) | Used to correct for variability in sample preparation and instrument response, mitigating matrix effects. | Should be a stable isotope-labeled version of the analyte, or a structurally similar analog. |
| High-Purity Solvents & Reagents | For mobile phases, sample preparation, and extraction. | Minimizes background interference and contamination in blank samples. |
| Solid Phase Extraction (SPE) Cartridges | A common sample preparation technique to clean up the sample and concentrate the analyte. | The selectivity of the sorbent is crucial for removing matrix interferences. |
In the rigorous world of analytical method validation, the concepts of specificity and selectivity form a foundational pillar for ensuring the accuracy and reliability of chromatographic methods. Specificity, the ideal capability of a method to confirm the identity of an analyte unequivocally in the presence of other components, represents the ultimate goal for confirmatory assays [41]. In practice, this is often demonstrated through the achievement of baseline resolution in chromatographic separations. Selectivity, the practical capability to differentiate an analyte from other substances like impurities or excipients, is a necessary and achievable standard, typically confirmed when the resolution between an analyte and any interfering peak is greater than 2.0 [41]. This whitepaper provides an in-depth technical guide for researchers and drug development professionals on the theory and practical strategies to achieve baseline resolution, thereby ensuring methods are not only selective but approach the gold standard of specificity required for robust analytical validation.
Chromatographic resolution ((R_s)) is a quantitative measure of the separation between two adjacent peaks [43] [44]. It is mathematically defined as:
[ Rs = \frac{2(t{R2} - t{R1})}{w1 + w_2} ]
where (t{R2}) and (t{R1}) are the retention times of the two peaks, and (w1) and (w2) are their respective baseline widths [44] [45].
The relationship between the calculated resolution value and the degree of peak separation is summarized in Table 1.
Table 1: Resolution Values and Their Practical Implications for Quantitation
| Resolution (Rₛ) | Degree of Separation | Theoretical Overlap | Implications for Quantitation |
|---|---|---|---|
| 0.8 | Partial overlap | ~5% mutual overlap | Potential for significant error if peak areas are unequal [43] |
| 1.0 | Partially resolved | ~2.2% mutual overlap | Minimum for "peak-to-peak" resolution; maximum 50% error possible with different detector responses [43] |
| 1.5 | Baseline resolution | ~0.3% mutual overlap | Considered sufficient for accurate quantitation; originally termed "99% baseline resolution" [43] [46] |
| 2.0 | Higher degree of separation | Near-complete | Often used as a benchmark for selectivity in method validation [41] |
From a method validation perspective, a method's selectivity is demonstrated by its ability to measure the analyte accurately in the presence of other components, which is practically achieved by ensuring adequate resolution between all peaks [41] [12]. Specificity is the ideal state where a method can unequivocally confirm the identity of an analyte, often represented by a chromatogram where only the target analyte elutes with no interference whatsoever [41]. For related substances testing, however, a method must be selectively powerful enough to separate and resolve all impurities from the main peak and from each other, meaning it should not be "too specific" [41].
The Fundamental Equation of Resolution
The practical optimization of resolution is guided by a fundamental equation that deconstructs (R_s) into three independent parameters: efficiency ((N)), selectivity ((\alpha)), and retention ((k)) [47] [48]:
[ R_s = \frac{\sqrt{N}}{4} \times \frac{\alpha - 1}{\alpha} \times \frac{k}{k + 1} ]
This equation provides a systematic framework for method development. The following diagram illustrates the logical decision process for optimizing each parameter.
Experimental Protocols for Optimizing Resolution
Protocol 1: Optimizing Mobile Phase for Selectivity (α)
Changing the mobile phase composition is often the most powerful approach for improving selectivity and achieving baseline resolution [47].
Detailed Methodology:
- Initial Conditions: Begin with a standard reversed-phase system (e.g., C18 column, 50:50 Acetonitrile:Water) and note the critical pair that is not resolved.
- Change Organic Modifier: Replace the organic solvent while maintaining equivalent elution strength. For example, if using 50% Acetonitrile, switch to approximately 57% Methanol or 35% Tetrahydrofuran (THF) using solvent strength relationships [47]. Analyze the sample and observe changes in the elution order and resolution of the critical pair.
- Adjust pH (for ionizable compounds): Prepare mobile phases with buffers (e.g., 25 mM phosphate or formate) at different pH values. For acidic compounds (pKa <5), use a low pH (e.g., 2.5-3.5) to suppress ionization and increase retention. For basic compounds (pKa >7), use a higher pH (e.g., 7-10, compatible with the column) [48]. Analyze the sample at each pH, noting the impact on the resolution of the critical pair.
- Fine-tune with Mixed Modifiers: Experiment with mixtures of two organic modifiers (e.g., Acetonitrile and Methanol) in varying ratios to fine-tune selectivity [47].
Protocol 2: Enhancing Efficiency (N) with Column and Temperature
Increasing the plate number sharpens peaks, which directly improves resolution [47] [48].
Detailed Methodology:
- Column Particle Size:
- Equilibrate two columns of the same dimensions and chemistry but different particle sizes (e.g., 5 µm and 3 µm or 1.7 µm).
- Inject the sample onto both columns under identical mobile phase and flow rate conditions.
- Calculate the efficiency (N) and resolution (Rₛ) for the critical pair on each column. Smaller particles will yield higher N and Rₛ, though at a higher system backpressure [47] [48].
- Column Length:
- Perform a separation on a standard 50 mm or 100 mm column.
- Switch to a longer column of the same chemistry (e.g., 150 mm or 250 mm). Adjust the flow rate proportionally to maintain a similar linear velocity and analysis time if desired (e.g., double the flow rate when doubling the column length) [47].
- Compare the resolution and peak capacity. The longer column will generate more theoretical plates, improving the separation of complex mixtures.
- Column Temperature:
- Set the column compartment to a starting temperature (e.g., 30°C) and perform an analysis.
- Incrementally increase the temperature (e.g., to 40°C, 50°C, 60°C) and analyze the sample at each step.
- Record the resolution of the critical pair and the system backpressure. Higher temperatures reduce mobile phase viscosity, increasing efficiency and often reducing backpressure, but may also alter selectivity for ionizable compounds [47] [49].
Protocol 3: System Suitability and Peak Shape Analysis
For a method to be valid, it must demonstrate consistent performance through system suitability testing, which includes resolution [12].
Detailed Methodology:
- Prepare a Test Solution: Create a solution containing the analyte and its closest eluting impurity or a standard that contains a critical pair of peaks.
- Establish Resolution Criteria: Set a system suitability requirement for resolution, typically Rₛ > 1.5 between the critical pair [43] [46].
- Perform Replicate Injections: Inject the test solution a minimum of five times.
- Calculate Key Parameters:
- Resolution (Rₛ): Calculate for the critical pair as per the equation in Section 1.
- Tailing Factor (T): Measure to ensure peak symmetry ((T \leq 2.0) is typical).
- Repeatability: Calculate the relative standard deviation (RSD%) of peak areas and retention times for the analyte (typically RSD ≤ 1.0% for retention time).
- The method is considered suitable only if all predetermined criteria for resolution, tailing, and repeatability are met before sample analysis.
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key solutions and materials required for developing and executing a robust chromatographic method with baseline resolution.
Table 2: Essential Research Reagent Solutions for Critical Separations
| Item | Function / Purpose | Technical Considerations |
|---|---|---|
| HPLC/UHPLC Column Suite | The stationary phase is the heart of the separation. | Maintain a small library of columns (e.g., C18, C8, Phenyl, HILIC) with different particle sizes (1.7-5 µm) and lengths (50-250 mm) to screen for selectivity and efficiency [47] [48]. |
| HPLC-Grade Organic Solvents | Primary mobile phase components for reversed-phase chromatography. | Acetonitrile (most common), Methanol, and Tetrahydrofuran (THF). Each offers different selectivity and should be on hand for method development [47]. |
| Buffer Salts and Additives | Control pH and ionic strength to manipulate retention and selectivity of ionizable compounds. | Ammonium formate/acetate, Potassium phosphate, Trifluoroacetic Acid (TFA), Formic Acid. Use volatile buffers for LC-MS compatibility [49] [48]. |
| System Suitability Test Mix | Verify column performance and instrument calibration before analytical runs. | A mixture of standard compounds (e.g., uracil for (t_0), and other probes for efficiency, tailing, and resolution) to confirm the system is within specified parameters [12]. |
| Reference Standards and APIs | For peak identification, calibration, and method validation. | Highly purified characterized materials of the Active Pharmaceutical Ingredient (API) and known impurities/degradants to establish identity, specificity, and selectivity [12]. |
Achieving baseline resolution is a critical objective in the development of robust, reliable chromatographic methods for drug development. It serves as the practical bridge between the concepts of selectivity—a method's practical capability to distinguish an analyte from interferents—and specificity, the ideal state of unequivocal identification. By systematically applying the theoretical principles and experimental protocols outlined in this guide, scientists can effectively optimize the three levers of chromatographic resolution: efficiency, selectivity, and retention. This systematic approach ensures that analytical methods are not only capable of accurate quantitation but also meet the rigorous validation requirements of regulatory standards, thereby safeguarding product quality and patient safety.
In the realm of pharmaceutical development, the validation of analytical methods is paramount to ensuring drug safety, efficacy, and quality. Within this framework, the concepts of specificity and selectivity represent critical validation parameters that determine an method's ability to accurately measure an analyte in the presence of potential interferents [50]. While often used interchangeably, these terms carry distinct meanings: specificity refers to the ability to unequivocally assess the analyte in the presence of components that may be expected to be present, while selectivity refers to the ability to distinguish the analyte from other analytes in the mixture [50] [51]. This case study examines how these principles are applied through High-Performance Liquid Chromatography (HPLC) and Liquid Chromatography-Mass Spectrometry (LC-MS) methodologies in modern drug development, highlighting their complementary roles through experimental data and regulatory considerations.
Fundamental Principles and Technological Differences
HPLC and LC-MS Core Technologies
High-Performance Liquid Chromatography (HPLC) is a chromatographic technique that separates compounds based on their differential interactions with a stationary phase and a mobile phase pumped under high pressure [52] [53]. In HPLC, sample components are separated as they travel through a column packed with fine particles, with compounds interacting differently with the stationary phase and thus eluting at distinct retention times [53]. Detection is typically achieved through ultraviolet-visible (UV-Vis), fluorescence, or other detectors that measure physical properties of the compounds [52] [53].
Liquid Chromatography-Mass Spectrometry (LC-MS) combines the separation capabilities of HPLC with the mass analysis power of mass spectrometry [52] [54]. After chromatographic separation, compounds are ionized (commonly through electrospray ionization), and the mass spectrometer measures their mass-to-charge ratio (m/z) [54] [53]. This hybrid approach provides both separation and structural identification capabilities in a single analytical platform [52].
Comparative Advantages and Limitations
The fundamental differences between HPLC and LC-MS technologies translate to distinct advantages for specific applications in drug development:
Table 1: Comparison of HPLC and LC-MS Characteristics in Drug Development
| Parameter | HPLC | LC-MS |
|---|---|---|
| Principle of Detection | Physical properties (e.g., UV absorption, fluorescence) [52] | Mass-to-charge ratio of ionized compounds [52] |
| Specificity & Selectivity | Good with optimal separation; may struggle with co-eluting peaks [50] | Superior; can distinguish compounds by mass even with co-elution [52] [55] |
| Sensitivity | Good with advanced detectors (e.g., fluorescence) [56] | Excellent; capable of detecting trace compounds at picogram levels [52] [54] |
| Sample Preparation | Typically simpler (dilution, filtration) [52] | May require additional steps for matrix compatibility [52] |
| Ideal Applications | Routine analysis of known compounds, quality control [52] | Complex samples, unknown identification, metabolite profiling [55] [57] |
Case Study: Method Development and Validation for Specificity and Selectivity
Establishing Specificity in HPLC Methods
For stability-indicating HPLC methods, specificity must be demonstrated through forced degradation studies that investigate main degradative pathways [50]. These studies provide samples with sufficient degradation products to evaluate the method's ability to separate the active pharmaceutical ingredient (API) from process impurities and degradation products [50].
In a case study developing a stability-indicating HPLC method for Tonabersat, researchers validated specificity by demonstrating baseline separation of the API from all potential impurities and degradation products [58]. Similarly, for sotalol hydrochloride, specificity was confirmed through forced degradation under acidic, alkaline, oxidative, photolytic, and thermal stress conditions, showing resolution >3.0 between all adjacent peaks and no interference from blank solutions [51].
A key approach to demonstrating specificity involves peak purity assessment using photodiode array (PDA) detectors, which confirms that analyte peaks are not contaminated with co-eluting impurities [50]. When developing a method for cardiovascular drugs in human plasma, researchers used dual UV and fluorescence detection to confirm specificity, with optimized excitation/emission wavelengths for each compound to ensure selective detection [56].
Enhanced Selectivity Through LC-MS Approaches
LC-MS provides inherent selectivity advantages through mass-based discrimination. In a study quantifying the mTOR inhibitor AC1LPSZG in rat plasma, researchers employed multiple reaction monitoring (MRM) to monitor specific transitions from precursor to product ions, providing unparalleled selectivity even in complex biological matrices [55]. The method achieved a linear range of 10-5000 ng/mL with precision and accuracy within ±15%, demonstrating robust selectivity for pharmacokinetic studies [55].
Another case study analyzing Andrographis paniculata extract in human plasma and urine developed a highly selective LC-MS/MS method that simultaneously quantified four major diterpenoids and their phase II metabolites [57]. The method's selectivity enabled detection at sub-nanogram per milliliter levels, overcoming limitations of previous methods that had restricted detectable plasma levels during the elimination phase [57].
Experimental Protocols for Specificity and Selectivity Assessment
Forced Degradation Protocol for HPLC Specificity (Based on ICH Guidelines) [50] [51]:
Prepare sample solutions under various stress conditions:
- Acidic hydrolysis: 1M HCl at room temperature or elevated temperature
- Alkaline hydrolysis: 1M NaOH at room temperature or elevated temperature
- Oxidative degradation: 3% H₂O₂ at room temperature
- Thermal degradation: Heat at 105°C for defined periods
- Photolytic degradation: Expose to UV light at specified intensity
Analyze stressed samples alongside untreated controls and placebo formulations
Evaluate chromatographic separation to ensure:
LC-MS Selectivity Validation Protocol [55] [57]:
Analyze blank samples from at least six different sources to confirm absence of interference
Confirm specificity of MRM transitions by demonstrating:
- Consistent retention times across samples
- Signal-to-noise ratio >10:1 at the lower limit of quantification
- No cross-talk between MRM channels for multi-analyte methods
For metabolite identification, employ orthogonal techniques:
- Use different collision energies for fragmentation patterns
- Compare retention times and fragmentation with reference standards
- Apply liquid chromatography-quadrupole time-of-flight mass spectrometry (LC-QTOF/MS) for untargeted metabolite profiling [57]
Quantitative Method Validation Data
Method validation requires demonstrating that analytical procedures are suitable for their intended use. The following table summarizes typical validation parameters and acceptance criteria for HPLC and LC-MS methods in pharmaceutical analysis:
Table 2: Method Validation Parameters and Acceptance Criteria for HPLC and LC-MS Methods
| Validation Parameter | HPLC Acceptance Criteria | LC-MS Acceptance Criteria | Regulatory Reference |
|---|---|---|---|
| Specificity | No interference from blank, placebo, or degradation products; Resolution >2.0 between critical pairs [50] | No interference in blank matrix; Specific MRM transitions for each analyte [55] | ICH Q2(R1) [50] |
| Accuracy | Recovery 98-102% for assay, 90-107% for impurities [50] | Recovery 85-115% with RSD <15% [55] | ICH Q2(R1) [50] |
| Precision | RSD <2% for assay, <5-10% for impurities [50] | RSD <15% at LLOQ, <10% at other levels [57] | ICH Q2(R1) [50] |
| Linearity | r² ≥ 0.998 over specified range [59] | r² ≥ 0.99 over specified range [55] | ICH Q2(R1) [50] |
| Range | 80-120% of test concentration for assay; LOQ-120% of specification for impurities [50] | LLOQ to ULOQ covering expected concentrations [55] | ICH Q2(R1) [50] |
| LOD/LOQ | Signal-to-noise ratio 3:1 for LOD, 10:1 for LOQ [59] | Signal-to-noise ratio 3:1 for LOD, 10:1 for LOQ [55] | USP <1225> [50] |
Experimental Workflows and Signaling Pathways
The following workflow diagrams illustrate the logical relationships and experimental processes for HPLC and LC-MS method development in drug development contexts:
Diagram 1: HPLC Method Development Workflow
Diagram 2: LC-MS/MS Method Development Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful method development requires carefully selected reagents and materials. The following table outlines key components for HPLC and LC-MS applications:
Table 3: Essential Research Reagent Solutions for HPLC and LC-MS Method Development
| Reagent/Material | Function/Purpose | Application Examples |
|---|---|---|
| C18/C8 Columns | Reverse-phase separation medium; different selectivity for various compounds [59] | Pharmaceutical impurities, stability testing [50] [59] |
| Tetrabutylammonium Salts | Ion-pairing agents for separation of ionic compounds [59] | Simultaneous analysis of ionic and non-ionic compounds [59] |
| Mass Spectrometry-Compatible Buffers | Volatile buffers (ammonium formate/acetate) that don't interfere with ionization [55] | LC-MS methods for biological samples [55] [57] |
| Protein Precipitation Reagents | Organic solvents (acetonitrile, methanol) for removing proteins from biological samples [55] | Bioanalytical sample preparation for plasma/serum [55] [56] |
| LLE Solvents | Organic solvents (diethyl ether, dichloromethane) for extracting analytes from aqueous matrices [56] | Sample clean-up and concentration for trace analysis [56] |
| Stable Isotope-Labeled Internal Standards | Correction for matrix effects and recovery variations in quantitative LC-MS [55] | Bioanalytical method for pharmacokinetic studies [55] [57] |
The complementary application of HPLC and LC-MS methodologies provides a comprehensive framework for addressing diverse analytical challenges in drug development. HPLC remains the workhorse for routine analysis, stability testing, and quality control where specificity is achieved through chromatographic separation [52] [50]. In contrast, LC-MS offers enhanced selectivity through mass-based discrimination, making it indispensable for complex matrices, metabolite identification, and trace-level quantification [55] [54] [57]. The strategic selection between these techniques, or their orthogonal application, should be guided by the specific analytical requirements, with method validation rigorously demonstrating the required specificity and selectivity for the intended purpose. As drug development advances toward more complex molecules and lower dosage regimens, the integration of these technologies will continue to evolve, maintaining their critical role in ensuring pharmaceutical product quality and patient safety.
Solving Common Challenges: Interferences, Co-elution, and Method Enhancement
In analytical method validation, the accurate quantification of an analyte is paramount. This accuracy is directly threatened by analytical interference, defined as the effect of a substance that causes the measured concentration of an analyte to differ from its true value [60]. Managing interference is fundamentally linked to two critical, yet distinct, validation parameters: specificity and selectivity.
Specificity is formally defined as the "ability to assess unequivocally the analyte in the presence of components which may be expected to be present" [1]. It describes a method's power to identify a single key—the target analyte—within a complex bunch, without necessarily needing to identify all the other keys present [1] [11]. Selectivity, while often used interchangeably, is a broader concept. It refers to the ability of a method to differentiate and quantify multiple analytes in the presence of other components in the sample [1] [11]. In essence, while a specific method finds the one right key, a selective method can identify all keys in the bunch. A robust analytical method must be designed to maximize both attributes to ensure results are reliable and unequivocal, forming the core thesis of effective method validation.
Interferences in analytical chemistry can originate from a myriad of sources and manifest in different ways, impacting both the selectivity and specificity of a method. Understanding this taxonomy is the first step toward effective mitigation.
Table 1: Common Sources of Analytical Interference
| Source Category | Examples | Primary Impact |
|---|---|---|
| Patient/Treatment Related | Common prescription drugs, over-the-counter medications, dietary supplements, parenteral nutrition, plasma expanders [60]. | Specificity, Selectivity |
| Sample Matrix | Hemolysis, icterus, lipemia, proteins, phospholipids [60] [61]. | Matrix Effects |
| Sample Handling & Preparation | Anticoagulants (e.g., EDTA, heparin), preservatives, stabilizers, contaminants from collection tubes (stopper leachables, serum separators), hand creams [60]. | Specificity, Matrix Effects |
| Structurally Related Compounds | Impurities, degradation products, metabolites, isobaric compounds, deuterium-labeled internal standards with isotope effects [1] [60] [61]. | Specificity, Selectivity |
The manifestation of these interferents can be broadly classified into two types:
- Analytical Interferences: These cause a direct alteration in the measured signal of the analyte. A common example is coelution in chromatographic techniques, where an interferent is not sufficiently separated from the analyte of interest, leading to a combined and inaccurate signal [62]. Isobaric interferences in mass spectrometry, where a compound shares the same mass-to-charge ratio as the analyte, are another significant challenge, particularly if they also coelute [60].
- Matrix Effects: Predominantly observed in mass spectrometry, matrix effects occur when substances in the sample alter the ionization efficiency of the analyte, typically causing signal suppression or, less commonly, signal enhancement [60] [61]. This does not necessarily involve a direct signal from the interferent but rather an indirect impact on the analyte's detectability.
Figure 1: A taxonomy of common interference types in analytical chemistry, showing the two primary categories and their sub-types.
Strategies for Interference Mitigation
A multi-pronged approach leveraging sample preparation, instrumental separation, and detection specificity is required to mitigate interferences and enhance method robustness.
Sample Preparation Techniques
Sample preparation is a primary defense for purifying and concentrating the analyte while removing potential interferents.
- Solid-Phase Extraction (SPE): This technique uses cartridges with various sorbents to trap analytes and wash away interferents. It is highly effective for pre-concentrating analytes from dilute aqueous samples, like environmental water, and for desalting [61].
- Liquid-Liquid Extraction (LLE): This method leverages the differential solubility of analytes and interferents between two immiscible solvents (e.g., organic and aqueous phases) for separation and clean-up [61].
- Protein Precipitation: Commonly used for biological samples like plasma or serum, this technique adds an organic solvent or acid to precipitate proteins, which are then removed by centrifugation, reducing matrix complexity [61].
- Derivatization: This process chemically modifies the analyte to make it more amenable to analysis by, for example, increasing its volatility for Gas Chromatography (GC) or improving its detectability [61]. It can also be used to "trap" reactive analytes like formaldehyde to prevent loss [61].
- Solid-Phase Microextraction (SPME): A solvent-free technique where a coated fiber extracts volatiles or non-volatiles from a sample headspace or via direct immersion, ideal for on-site sampling [61].
Chromatographic Separation
Liquid Chromatography (LC) is a powerful tool for achieving separation selectivity [1]. A well-optimized method can physically separate the analyte from potential interferents before they reach the detector.
- Column Chemistry: Careful selection of the stationary phase (e.g., C18, phenyl, HILIC) is crucial as it dictates the chemical interactions that resolve compounds [60].
- Mobile Phase and Gradient Elution: Manipulating the composition of the mobile phase and its gradient profile over time allows the laboratory to maneuver analytes into regions of the chromatogram free from matrix effects and coeluting interferents [60].
Mass Spectrometric Detection and Selectivity
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) provides an exceptional level of analytical selectivity through a combination of physical separation and mass-based detection.
- Selected/Multiple Reaction Monitoring (SRM/MRM): This is the cornerstone of selectivity in LC-MS/MS. The first quadrupole (MS1) selects the precursor ion of the analyte. This ion is fragmented in the collision cell, and the second quadrupole (MS2) selects one or more unique product ions. This two-stage mass filtering provides a high degree of certainty in analyte identification [60].
- Ion Ratio Monitoring: The ratio of intensities between two or more product ions for a given analyte is a fixed characteristic. Monitoring this ratio during routine analysis serves as a key data quality metric; a deviation outside pre-defined limits signals potential interference [60].
Figure 2: The LC-MS/MS workflow for interference mitigation, showing how chromatographic and mass-based selectivity remove different interferents.
The Role of Internal Standards
Internal standards (IS) are critical for compensating for variability during sample preparation and analysis, particularly matrix effects.
- Stable Isotope-Labeled Internal Standards (SIL-IS): These are the gold standard, especially for MS-based assays. An isotopically labeled version of the analyte (e.g., with ²H, ¹³C, ¹⁵N) is added at the beginning of sample preparation. It co-elutes with the native analyte and experiences nearly identical ionization effects, allowing for accurate correction [60] [61].
- Deuterium Isotope Effect: A potential pitfall with deuterated standards is that they can exhibit slightly different chromatographic retention times compared to the native analyte, which can compromise their ability to correct for matrix effects if they do not perfectly co-elute. Where possible, ¹³C or ¹⁵N labeled IS are preferred to avoid this issue [61].
Table 2: Key Research Reagent Solutions for Interference Mitigation
| Reagent / Material | Function | Key Consideration |
|---|---|---|
| Stable Isotope-Labeled Internal Standard (SIL-IS) | Compensates for analyte loss during prep and matrix effects during ionization; essential for quantification [60] [61]. | Should be added early in sample prep; must co-elute with analyte; 13C/15N labels preferred over deuterium to avoid retention time shifts [61]. |
| SPE Sorbents | Selectively binds analyte or interferents for sample clean-up and pre-concentration [61]. | Choice of sorbent (e.g., C18, ion-exchange, mixed-mode) is dictated by analyte and matrix physicochemical properties. |
| Derivatization Reagents | Chemically modifies analyte to improve volatility (for GC), detectability, or stability [61]. | Reagent must be specific to the analyte's functional group; process should be efficient and reproducible. |
| LC Mobile Phase Additives | Modifies chromatographic retention and selectivity to achieve separation of interferents [60]. | Must be MS-compatible (e.g., volatile acids, buffers); composition and pH critically impact separation. |
Experimental Protocols for Interference Testing
Rigorous interference testing is a non-negotiable component of method development and validation. The following protocols provide a framework for this assessment.
Testing for Specific Interferences
This protocol evaluates the effect of known, specific substances on the assay.
- Preparation of Pools: Prepare a base pool of the sample matrix (e.g., plasma, urine) with a known concentration of the target analyte.
- Spiking of Interferents: Following guidelines such as CLSI EP7-A2, spike the highest concentration expected in vivo of the potential interferent (e.g., a common drug, bilirubin for icterus) into the test pool. A control pool is spiked with an equal volume of solvent [60].
- Analysis and Calculation: Analyze the test and control pools with adequate replication within the same analytical run. Calculate the percentage bias using the formula:
Bias (%) = [(Mean Concentration of Test Pool - Mean Concentration of Control Pool) / Mean Concentration of Control Pool] × 100 - Interpretation: A bias that exceeds pre-defined clinical acceptability limits (e.g., ±15%) indicates clinically significant interference. For such interferents, further testing at different concentrations is warranted to determine the threshold for interference [60].
Post-Column Infusion for Matrix Effects
This qualitative experiment visualizes regions of ion suppression or enhancement in the chromatogram.
- Setup: A solution of the analyte (or its isotopically labeled internal standard) is continuously infused via a post-column T-connector into the LC effluent flowing to the MS.
- Injection: A blank matrix sample (e.g., drug-free plasma) extracted using the normal sample preparation protocol is injected onto the LC column and the chromatographic gradient is run.
- Visualization: The signal of the infused analyte is monitored. A steady signal indicates no matrix effects. A dip in the signal (suppression) or a peak (enhancement) indicates the elution of matrix components that affect ionization [60].
- Application: The results guide method development. The LC gradient or sample clean-up procedure can be modified to ensure the analyte elutes in a region of minimal matrix effect [60].
Quantitative Matrix Effect Experiment
This protocol provides a numerical value for the extent of matrix effects.
- Sample Sets: Prepare two sets of samples:
- Set A (Extracted Matrix): Spike the analyte into several (e.g., 6 or more) lots of blank matrix after they have undergone the complete sample extraction process.
- Set B (Neat Solution): Spike the same amount of analyte into a pure solvent.
- Analysis: Analyze all samples and record the peak areas of the analyte (and internal standard, if used).
- Calculation: Calculate the matrix effect (ME) for each matrix lot.
- Without IS:
ME (%) = (Mean Peak Area of Set A / Mean Peak Area of Set B) × 100 - With IS: Calculate the response factor (RF = Analyte Peak Area / IS Peak Area) for each set, then
ME (%) = (Mean RF of Set A / Mean RF of Set B) × 100
- Without IS:
- Interpretation: A value of 100% indicates no matrix effect. Values <100% indicate ion suppression, and >100% indicate enhancement. A coefficient of variation (CV) of the ME across different matrix lots of >15% is typically indicative of a significant and variable matrix effect that must be addressed [60].
In the context of analytical method validation, the journey from a non-selective to a highly specific and selective method is the path to reliability. Interference is an ever-present challenge, but it is not insurmountable. A systematic approach that combines an understanding of the sample matrix, judicious application of sample preparation techniques, optimization of chromatographic separation, and exploitation of the intrinsic selectivity of mass spectrometric detection, provides a robust framework for its identification and mitigation. The experiments outlined herein are not merely regulatory checkboxes but are fundamental practices that ensure the accuracy and precision of analytical data. By rigorously challenging a method with potential interferents during development and implementing ongoing quality controls, researchers and drug development professionals can confidently generate data that supports the safety and efficacy of pharmaceutical products.
Strategies for Resolving Co-elution and Overlapping Peaks in Chromatography
Chromatographic co-elution, the phenomenon where two or more compounds exit the chromatography column simultaneously, represents a critical challenge in analytical chemistry, particularly in pharmaceutical development and complex mixture analysis. This in-depth technical guide examines systematic strategies for detecting and resolving overlapping peaks, framed within the crucial context of specificity and selectivity in analytical method validation. Effective resolution of co-elution is fundamental to developing methods that can assess unequivocally the analyte in the presence of components which may be expected to be present—the very definition of specificity according to ICH Q2(R1) guidelines [63]. This guide provides researchers and drug development professionals with both theoretical foundations and practical experimental protocols to address this pervasive analytical challenge, ensuring data integrity and regulatory compliance.
Co-elution occurs when two peaks exit the chromatography column at nearly the same time, compromising our ability to properly identify and quantify individual compounds [64]. In a system fundamentally designed for separation, co-elution represents its "Achilles' heel"—invalidating results until resolved [64]. The problem is particularly prevalent in the analysis of complex biological mixtures where metabolites with similar chromatographic properties coexist [65]. In pharmaceutical contexts, unresolved peaks can lead to inaccurate potency assessments, incomplete impurity profiling, and flawed stability studies, ultimately jeopardizing drug safety and efficacy profiles.
The relationship between co-elution resolution and method validation parameters is inseparable. Specificity focuses on the method's ability to identify only the target analyte unequivocally, while selectivity involves distinguishing the analyte from other components in the sample [63]. A common misconception is that these terms are interchangeable; however, selectivity represents a broader capability to differentiate multiple components, while specificity targets exclusive identification [63]. Understanding this distinction is crucial when developing strategies to resolve co-elution, as approaches may prioritize one characteristic over the other depending on the analytical context.
Theoretical Foundation of Chromatographic Resolution
The Resolution Equation
The fundamental equation governing chromatographic separation provides a mathematical framework for understanding co-elution and systematically addressing it:
[ Rs = \frac{1}{4} \sqrt{N} \times \frac{\alpha - 1}{\alpha} \times \frac{k2}{1 + k_{avg}} ]
Where:
- (R_s) = resolution between two peaks
- (N) = column efficiency (theoretical plate count)
- (\alpha) = selectivity (relative retention ratio)
- (k) = capacity factor (retention factor) [47]
This equation reveals that resolution depends on three independent factors: efficiency (N), selectivity (α), and retention (k). The most powerful approach to improving resolution involves increasing α (selectivity), as even small changes can dramatically impact separation [47]. Understanding and manipulating these parameters forms the basis of all systematic approaches to resolving co-elution.
Specificity vs. Selectivity in Method Validation
In analytical method validation, understanding the distinction between specificity and selectivity is crucial:
- Specificity: The ability to assess unequivocally the analyte in the presence of components which may be expected to be present [63]. This focuses on identifying only the target analyte.
- Selectivity: The ability to differentiate the analyte(s) of interest from endogenous components in the matrix or other components in the sample [63]. This involves distinguishing multiple substances.
Few analytical methods are truly 100% specific, as most have some level of cross-reactivity or interference [63]. This reality makes selectivity often more valuable in real-world applications involving complex mixtures. When resolving co-elution, the goal is to enhance selectivity to achieve effective specificity for the intended analytical purpose.
Detection and Confirmation of Co-elution
Visual Indicators of Peak Overlap
Initial detection of co-elution often begins with visual inspection of chromatograms. Key indicators include:
- Shoulders on peaks: Sudden discontinuities in peak shape that may indicate two compounds exiting simultaneously [64]
- Asymmetric peak shapes: Deviations from normal Gaussian distribution
- Unexpected peak broadening: Wider than expected peaks given column parameters
- Baseline disturbances: Anomalies before, during, or after peak elution
It's important to distinguish between a tail (a gradual exponential decline) and a shoulder (a sudden discontinuity), as the latter more strongly suggests co-elution [64]. However, perfect co-elution with no obvious distortion can occur, requiring more sophisticated detection methods.
Detector-Based Confirmation Methods
Table 1: Detector-Based Approaches for Confirming Co-elution
| Detection Method | Principle of Operation | Experimental Protocol | Advantages |
|---|---|---|---|
| Diode Array Detector (DAD/PDA) | Collects ~100 UV spectra across a single peak [64] | Compare spectra from different points (up-slope, apex, down-slope) of the peak | Non-destructive; provides spectral evidence of purity |
| Mass Spectrometry (LC-MS/GC-MS) | Analyzes mass-to-charge ratio and fragmentation patterns [66] | Create Extracted Ion Chromatograms (EICs) for specific m/z values; match spectra to libraries | Provides molecular weight and structural information; high sensitivity |
| Peak Purity Analysis | Algorithms compare spectra across the peak | Software-based assessment of spectral homogeneity | Automated; provides numerical purity indices |
Diode array detectors are particularly valuable for peak purity analysis. If spectra collected across a single peak are identical, you likely have a pure compound; if they differ, the system flags potential co-elution [64]. With mass spectrometry, the same concept applies—taking spectra along the peak and comparing them can reveal shifting profiles that indicate multiple compounds [66].
Experimental Strategies for Resolving Co-elution
Optimization of Chromatographic Parameters
Adjusting Retention (Capacity Factor k')
When co-elution occurs with low retention (k' < 1), peaks are flying through the system with the void volume [64].
Experimental Protocol:
- Weaken mobile phase: Reduce organic solvent concentration in reversed-phase HPLC
- Adjust gradient profile: Implement shallower gradients to increase separation time
- Target optimal range: Aim for k' between 1 and 5 for balanced analysis time and resolution [64]
Example: For a method using 70% acetonitrile where co-elution occurs, systematically reduce organic content to 60%, 50%, or 40% while monitoring resolution of critical pairs.
Enhancing Selectivity (α)
Selectivity reflects how differently analytes interact with the stationary phase and represents the most powerful approach to resolving co-elution [47].
Experimental Protocol:
- Change organic modifier: Switch between acetonitrile, methanol, or tetrahydrofuran using solvent strength relationships to maintain similar retention times [47]
- Adjust mobile phase pH: For ionizable compounds, modify pH to alter ionization states and retention (typically 2 units away from pKa for maximum effect)
- Use alternative columns: Select different stationary phase chemistries (C8, C18, phenyl, cyano, HILIC) [64] [67]
Table 2: Solvent Strength Relationships for Modifier Replacement
| Original Condition | Alternative 1 (Methanol) | Alternative 2 (THF) |
|---|---|---|
| 50% Acetonitrile | 57% Methanol | 35% Tetrahydrofuran |
| 60% Acetonitrile | 68% Methanol | 42% Tetrahydrofuran |
| 40% Acetonitrile | 46% Methanol | 28% Tetrahydrofuran |
Data adapted from solvent strength relationships [47]
Improving Efficiency (N)
Column efficiency measures peak sharpness and can be enhanced through several approaches:
Experimental Protocol:
- Reduce particle size: Columns with smaller particles produce higher plate numbers for sharper peaks [47]
- Increase column length: Longer columns provide more theoretical plates but increase run times and backpressure
- Optimize temperature: Higher temperatures reduce mobile phase viscosity and increase diffusion rates (40-60°C for small molecules; 60-90°C for large molecules) [47]
- Adjust flow rate: Identify optimal flow rate for maximum efficiency using van Deemter curves
Example: Figure 1 from the literature shows resolution increased from approximately 0.8 to 1.25 by using a column with smaller particles (e.g., transitioning from 3.0 μm to 2.7 μm or 1.7 μm particles) while maintaining the same column dimensions [47].
Advanced Separation Techniques
Comprehensive Two-Dimensional Chromatography
For extremely complex samples, comprehensive two-dimensional liquid chromatography (LC×LC) provides significantly enhanced separation power [68]. This technique uses two different separation mechanisms (e.g., reversed-phase in the first dimension and HILIC in the second) to achieve peak capacities exceeding those of one-dimensional systems.
Experimental Considerations:
- Modulation technology: Active solvent modulators can address elution strength incompatibility between dimensions [68]
- Orthogonality: Select separation mechanisms with different retention mechanisms for maximum effectiveness
- Data analysis: Complex data requires specialized software and potentially feature clustering for interpretation [68]
Recent innovations include multi-2D LC×LC, where a six-way valve selects between different secondary dimensions (e.g., HILIC or RP) depending on the analysis time in the first dimension, significantly improving separation performance [68].
Computational Peak Deconvolution
When chemical separation proves insufficient, computational methods can mathematically resolve overlapping peaks:
Method 1: Clustering-Based Separation
- Procedure: Data normalization → baseline removal → retention time alignment → peak detection → clustering of convolved chromatogram fragments [65]
- Application: Suitable for large datasets with multiple samples; groups similar peaks by shape
Method 2: Functional Principal Component Analysis (FPCA)
- Procedure: Similar preprocessing followed by FPCA to detect sub-peaks with greatest variability [65]
- Advantage: Provides optimal multidimensional peak representation and assesses variability of individual compounds within same peaks across different chromatograms [65]
Both methods have been experimentally validated using metabolomic data from barley leaves under drought stress, demonstrating applicability to real-world biological samples [65].
The Scientist's Toolkit: Essential Materials and Reagents
Table 3: Research Reagent Solutions for Resolving Co-elution
| Tool Category | Specific Examples | Function/Purpose |
|---|---|---|
| Stationary Phases | C18, C8, Phenyl, Cyano, HILIC, Biphenyl, Amide, AR columns [64] | Alters selectivity through different chemical interactions with analytes |
| Organic Modifiers | Acetonitrile, Methanol, Tetrahydrofuran [47] | Changes solvent strength and selectivity; primary mobile phase components |
| Aqueous Buffers | Phosphate, acetate, ammonium formate, ammonium acetate | Controls pH and ionic strength to manipulate ionization of analytes |
| Column Hardware | Monodisperse particles (1.7-5μm), different lengths (30-250mm), varied diameters [67] | Provides different efficiency parameters and loading capacity |
| Detection | DAD/PDA, MS (QTOF, Orbitrap), ELSD/CAD, RID [67] [66] | Confirms peak purity and identity through spectral information |
| Sample Preparation | SPE cartridges, derivatization reagents, filtration devices [69] | Removes matrix interferents and concentrates analytes |
| Software Tools | ChemStation, Empower, ChromSwordAuto, S-Matrix Fusion QbD [69] | Automates method development and provides peak deconvolution algorithms |
Method Validation Considerations
When implementing strategies to resolve co-elution, method validation must confirm that approaches have successfully addressed the issues while maintaining overall method performance:
- Specificity/SELECTIVITY: Demonstrate baseline separation of critical pairs (resolution > 1.5 for compendial methods) [63]
- Accuracy: Confirm no interference in quantification of main analyte and impurities
- Precision: Verify consistent retention times and resolution across injections
- Robustness: Test method performance under deliberate variations of critical parameters (pH, temperature, mobile phase composition)
Documentation should clearly demonstrate the method's ability to distinguish the analyte from all potential impurities, degradation products, and matrix components. For pharmaceutical applications, forced degradation studies provide critical validation of method selectivity under stress conditions [67].
Resolving chromatographic co-elution requires a systematic approach grounded in the fundamental principles of the resolution equation. By methodically addressing retention, efficiency, and—most powerfully—selectivity, analysts can develop robust methods that meet validation requirements for specificity and selectivity. The strategies outlined in this guide, from basic parameter optimization to advanced computational and multidimensional approaches, provide researchers with a comprehensive toolkit for tackling this challenging analytical problem. As chromatographic applications continue to evolve toward more complex samples, these resolution strategies become increasingly essential for generating reliable, defensible analytical data in pharmaceutical development and beyond.
The continuing innovation in chromatographic technologies—including smaller particles, more diverse stationary phases, sophisticated two-dimensional systems, and artificial intelligence-driven method development—promises enhanced capabilities for addressing co-elution challenges in even the most complex matrices [68] [69].
Optimizing Sample Preparation to Improve Selectivity
In the framework of analytical method validation, the concepts of specificity and selectivity are fundamental, yet they are often differentiated by a key nuance. According to ICH Q2(R1) guidelines, specificity is "the ability to assess unequivocally the analyte in the presence of components which may be expected to be present." [1] It describes a method's ability to correctly identify and measure a single target analyte amidst potential interferents. A helpful analogy is finding a single, correct key that opens a lock from a large bunch of keys; the method identifies only the target without needing to recognize the others [1] [11].
Selectivity, while sometimes used interchangeably with specificity, carries a broader meaning. It refers to the ability of a method to differentiate and quantify multiple analytes of interest simultaneously within a complex sample, accurately distinguishing them from endogenous matrix components or other sample constituents [1]. In the key analogy, selectivity requires the identification of all keys in the bunch, not just the one that opens the lock [1] [11]. The International Union of Pure and Applied Chemistry (IUPAC) recommends the term "selectivity" for analytical chemistry, as it encompasses the method's capacity to respond to several different analytes [1]. This whitepaper focuses on optimizing sample preparation—a critical and controllable phase of analysis—to enhance this comprehensive capability of methods to ensure reliable, accurate, and unambiguous results in pharmaceutical development and other complex matrices.
Theoretical Foundation: Specificity vs. Selectivity
The distinction between specificity and selectivity is not merely semantic; it dictates the design, validation, and application of an analytical procedure. Specificity is often considered an absolute ideal—a property of a method that is exclusively responsive to one, and only one, analyte [1]. In practice, this is rarely fully achievable, which makes the concept of selectivity more practical and widely applicable. Selectivity is the degree to which a method can determine particular analytes in mixtures or matrices without interference from other components [1].
This distinction is operationally critical. For instance, in a chromatographic method for a drug substance, specificity might be demonstrated by showing that the active pharmaceutical ingredient (API) peak is pure and unaffected by the presence of excipients, impurities, or degradation products [24]. Selectivity, however, would be demonstrated by the method's ability to successfully resolve and individually quantify the API, all known impurities, and any degradation products that form under stress conditions, all within a single run [7]. The ultimate expression of selectivity in chromatography is a baseline resolution between the peaks of all analytes of interest [1].
Sample preparation serves as the first and one of the most powerful lines of defense in achieving high selectivity. A well-designed sample preparation protocol can selectively isolate the analytes of interest, remove or reduce the concentration of potential interferents, and present the analytes in a form compatible with the instrumental analysis, thereby significantly reducing the burden on the final separation and detection system.
Core Strategies for Selective Sample Preparation
Optimizing sample preparation involves choosing and fine-tuning techniques that leverage the unique physical and chemical properties of the target analytes to separate them from the sample matrix. The following core strategies are pivotal.
Liquid-Liquid Extraction (LLE)
Liquid-Liquid Extraction (LLE) is a foundational technique that separates compounds based on their relative solubility in two immiscible liquids, typically an aqueous phase and an organic solvent.
- Principle of Selectivity: The selectivity of LLE is governed by the partition coefficient (K) of an analyte, which is the ratio of its equilibrium concentrations in the organic and aqueous phases. By choosing solvents with different polarities and adjusting the pH of the aqueous phase to manipulate the ionization state of ionizable analytes, high selectivity can be achieved. For example, a basic drug can be selectively extracted into an organic solvent from an aqueous matrix by alkalizing the solution to suppress its ionization, thereby increasing its lipophilicity.
- Protocol Outline:
- Combine the liquid sample with a carefully selected immiscible organic solvent in a separation funnel.
- Adjust the pH of the aqueous phase to ensure target analytes are in their neutral form (e.g., use sodium hydroxide for basic compounds or hydrochloric acid for acidic compounds).
- Shake the mixture vigorously for a set time to achieve equilibrium.
- Allow the phases to separate completely.
- Drain and collect the organic layer containing the extracted analytes.
- The extract can be evaporated to dryness and reconstituted in a solvent compatible with the subsequent instrumental analysis (e.g., HPLC mobile phase).
Solid-Phase Extraction (SPE)
Solid-Phase Extraction (SPE) is a more versatile and efficient extraction technique that utilizes a cartridge packed with a solid sorbent to selectively retain analytes from a liquid sample as it passes through.
- Principle of Selectivity: Selectivity in SPE is determined by the chemical nature of the sorbent material and the composition of the solvents used for conditioning, loading, washing, and eluting. The wide range of available sorbents (e.g., reversed-phase C18 for hydrophobic interactions, cation-exchange for positively charged analytes, mixed-mode sorbents combining multiple mechanisms) allows for highly selective isolation. The step-wise solvent protocol is designed to retain the analytes while washing away interferences, followed by a selective elution of the purified analytes.
- Protocol Outline:
- Conditioning: Pass methanol through the SPE cartridge to wet the sorbent, followed by water or a buffer to create the appropriate environment for analyte retention.
- Loading: Apply the prepared sample to the cartridge. Analytes of interest are retained on the sorbent while some matrix components pass through.
- Washing: Pass a solvent with a weak elution strength through the cartridge to remove undesired matrix components without displacing the analytes.
- Elution: Apply a strong, selective solvent to disrupt the analyte-sorbent interaction and release the purified analytes into a collection tube.
- The eluate is often evaporated and reconstituted for analysis.
Protein Precipitation
Protein Precipitation is a simple and rapid sample preparation technique primarily used for biological fluids like plasma or serum.
- Principle of Selectivity: This technique relies on the denaturation and precipitation of proteins using an organic solvent (e.g., acetonitrile or methanol), acid, or salt. When centrifuged, the precipitated proteins form a pellet, leaving the analytes of interest in the clarified supernatant. While excellent for removing proteins, it is less selective than SPE or LLE for removing other small molecule interferences, which remain in the supernatant.
- Protocol Outline:
- Mix a volume of the biological sample (e.g., plasma) with a larger volume (typically 2-4x) of a precipitating solvent like acetonitrile.
- Vortex the mixture vigorously to ensure complete protein denaturation.
- Centrifuge the sample at high speed (e.g., 10,000 x g) for 10 minutes to pellet the precipitated proteins.
- Carefully collect the supernatant, which can be diluted or directly injected into an HPLC system.
The table below summarizes the key characteristics, advantages, and limitations of these core techniques.
Table 1: Comparison of Core Selective Sample Preparation Techniques
| Technique | Mechanism of Selectivity | Best For | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Liquid-Liquid Extraction (LLE) | Partition coefficient based on solubility and pH | Extracting non-polar to moderately polar analytes from aqueous matrices; high-volume samples. | Simple setup, high capacity, cost-effective. | Emulsion formation, large solvent volumes, automation can be difficult. |
| Solid-Phase Extraction (SPE) | Multiple interaction modes (hydrophobic, ionic, etc.) between analyte and sorbent | Complex matrices (biofluids, environmental samples); trace-level analysis; requires high purity. | High selectivity and clean-up, amenability to automation, concentration of analytes. | More complex method development, cartridge cost, potential for channeling. |
| Protein Precipitation | Physical removal of proteins via denaturation | Rapid processing of biological samples (plasma, serum) for macromolecule removal. | Extremely fast, simple, high recovery for many small molecules. | Limited selectivity for small molecules, matrix effects in LC-MS, dilution of analyte. |
Advanced and Emerging Techniques
For challenging applications requiring exceptional selectivity, advanced techniques offer enhanced capabilities.
- Immunoaffinity Extraction: This technique uses the highly specific binding between an antibody and its target antigen (analyte). Antibodies immobilized on a solid support can extract the analyte and its structurally similar analogues with extreme selectivity from very complex matrices. It is particularly valuable for quantifying biomarkers, hormones, and toxins at very low concentrations.
- Molecularly Imprinted Polymers (MIPs): MIPs are synthetic polymers with tailor-made recognition sites complementary to the target molecule in shape, size, and functional groups. They act as "artificial antibodies," offering high chemical and thermal stability at a lower cost than biological antibodies. They are used in SPE cartridges for the selective solid-phase extraction of specific analytes.
- Derivatization: This chemical technique modifies the analyte to alter its properties. It can be used to improve a method's selectivity by making the analyte more amenable to a specific detection method (e.g., adding a fluorophore for fluorescence detection) or to change its chromatographic behavior to resolve it from co-eluting interferences.
Validation and Analytical Performance
The success of any sample preparation optimization must be demonstrated through rigorous method validation, assessing key performance parameters as defined by ICH Q2(R1) and other guidelines [70] [24].
- Assessing Selectivity: The primary test for selectivity involves analyzing a minimum of six independent sources of the blank matrix (e.g., plasma from different donors) and comparing the chromatograms with those of the same matrix spiked with the analytes and potential interferents. The method is considered selective if there is no significant interference (e.g., <20% of the lower limit of quantitation for the analyte) at the retention times of the analytes [70] [7].
- Impact on Other Validation Parameters:
- Accuracy and Precision: Effective sample preparation minimizes matrix effects that can suppress or enhance analyte signal, thereby improving the accuracy (closeness to the true value) and precision (reproducibility) of the results [24].
- Linearity: A clean sample extract reduces the risk of detector saturation or non-linear response, supporting a wider linear dynamic range [24].
- Sensitivity: Efficient pre-concentration of analytes during sample preparation (as in SPE) directly lowers the Limit of Detection (LOD) and Limit of Quantitation (LOQ), enhancing the method's sensitivity for trace-level analysis [24].
Table 2: Key Analytical Validation Parameters and the Impact of Selective Sample Prep
| Validation Parameter | Definition | Role of Selective Sample Preparation |
|---|---|---|
| Selectivity/Specificity | Ability to measure analyte unequivocally amid components expected to be present [1]. | Primary enabler. Directly removes interfering substances from the sample matrix. |
| Accuracy | Closeness of agreement between the accepted reference value and the value found [24]. | Reduces matrix effects that cause bias (signal suppression/enhancement). |
| Precision (Repeatability & Intermediate Precision) | Closeness of agreement between a series of measurements from multiple sampling of the same homogeneous sample [24]. | Improves method robustness against variations in matrix composition, leading to more reproducible results. |
| Linearity | Ability of the method to obtain test results proportional to the concentration of the analyte [24]. | Prevents detector fouling and non-linearity caused by matrix components. |
| Limit of Quantitation (LOQ) | Lowest concentration of an analyte that can be quantified with acceptable precision and accuracy [24]. | Pre-concentration of analytes and reduction of background noise allow for lower, more reliable LOQs. |
| Robustness | Capacity of the method to remain unaffected by small, deliberate variations in method parameters [7]. | A cleaner sample extract makes the final instrumental analysis less susceptible to minor fluctuations. |
The Scientist's Toolkit: Essential Reagents and Materials
The following table details key research reagent solutions and materials essential for implementing selective sample preparation protocols.
Table 3: Essential Research Reagent Solutions for Selective Sample Preparation
| Item | Function in Selective Sample Prep |
|---|---|
| Solid-Phase Extraction (SPE) Cartridges | Contain the sorbent material (e.g., C18, Mixed-Mode, Ion-Exchange) that selectively retains analytes based on chemical interactions. The choice of sorbent is the primary determinant of selectivity in SPE [70]. |
| High-Purity Organic Solvents (e.g., Acetonitrile, Methanol) | Used in LLE, SPE (as eluents), and protein precipitation. Their purity is critical to prevent introduction of interfering contaminants. Acetonitrile is particularly effective for protein precipitation and is a common solvent in reversed-phase SPE [7]. |
| Buffers and pH Adjusters (e.g., Phosphate Buffers, Ammonium Acetate, HCl, NaOH) | Critical for controlling the ionization state of ionizable analytes in LLE and SPE. This allows for selective retention/elution by switching between charged and neutral forms [7]. |
| Derivatization Reagents | Chemicals that react with specific functional groups on the target analyte to alter its properties, improving its detectability (e.g., for fluorescence or MS detection) or chromatographic behavior to resolve it from interferents [24]. |
| Internal Standards (Stable Isotope-Labeled, SIL-IS) | Compounds, structurally identical to the analytes but labeled with heavy isotopes (e.g., Deuterium, C-13), added to the sample at the beginning of preparation. They correct for variability in recovery and matrix effects during analysis, significantly improving accuracy and precision [1]. |
Experimental Workflow and Protocol Design
A generalized, optimized workflow for developing and executing a selective sample preparation method is outlined below, integrating the techniques and principles discussed.
Diagram 1: Sample Preparation Optimization Workflow
Detailed Protocol for a Selective Solid-Phase Extraction (SPE) Method:
This protocol provides a step-by-step guide for a reversed-phase SPE procedure for a plasma sample.
- Objective: To selectively extract a hypothetical drug candidate and its major metabolite from human plasma for LC-MS/MS analysis.
- Materials:
- Mixed-mode C8/cation-exchange SPE cartridges (60 mg, 3 mL).
- HPLC-grade methanol, acetonitrile, and water.
- Ammonium acetate buffer (10 mM, pH 5.0).
- Elution solvent: 5% ammonium hydroxide in methanol.
- Stable Isotope-Labeled Internal Standards (SIL-IS) for the drug and metabolite.
- Centrifuge, vortex mixer, and a positive pressure SPE manifold.
- Procedure:
- Step 1: Sample Pre-Treatment. Thaw plasma samples on ice. Centrifuge at 10,000 x g for 5 minutes to pellet any particulates.
- Step 2: Add Internal Standard. Pipette 100 µL of plasma into a clean tube. Add 20 µL of the SIL-IS working solution. Vortex briefly to mix.
- Step 3: Condition SPE Cartridge. Load the cartridge on the manifold. Pass 2 mL of methanol through the cartridge, followed by 2 mL of ammonium acetate buffer. Do not let the sorbent dry out.
- Step 4: Load Sample. Apply the entire treated plasma sample (120 µL) to the conditioned cartridge at a slow, drop-wise flow rate (~1 mL/min).
- Step 5: Wash. Pass 2 mL of ammonium acetate buffer through the cartridge to remove salts and polar proteins. Follow with 1 mL of 20% methanol in water to remove less polar interferences.
- Step 6: Elute. Place a clean collection tube under the cartridge. Pass 1.5 mL of the 5% ammonium hydroxide in methanol elution solvent through the cartridge to release the basic drug, metabolite, and IS. Collect the entire eluate.
- Step 7: Post-Preparation Processing. Evaporate the eluate to dryness under a gentle stream of nitrogen at 40°C. Reconstitute the dry residue in 150 µL of the initial LC-MS/MS mobile phase. Vortex thoroughly and centrifuge before transferring to an autosampler vial for analysis.
Optimizing sample preparation is an indispensable strategy for achieving the high degree of selectivity demanded by modern analytical challenges, particularly in regulated environments like pharmaceutical development. By moving beyond the simplistic goal of mere analyte extraction to a focused strategy of selective isolation and matrix clean-up, scientists can directly enhance key validation parameters including accuracy, precision, and sensitivity. A deep understanding of the distinction between specificity and selectivity provides the necessary theoretical framework for this optimization. As analytical science continues to push toward lower detection limits and more complex matrices, the role of robust, selective, and efficient sample preparation will only grow in importance, serving as the critical foundation upon which reliable and defensible data is built.
Adjusting Chromatographic Parameters (pH, Column, Gradient) for Better Separation
In pharmaceutical analysis, the validation of analytical methods is foundational to ensuring product quality, safety, and efficacy. Within this framework, specificity and selectivity represent distinct but related validation parameters critical for method reliability. Specificity is the method's ability to measure the analyte accurately in the presence of potential interferents like impurities, degradants, or matrix components [7]. Selectivity, meanwhile, refers to the method's capacity to distinguish the analyte from other substances in the sample based on chromatographic separation [71]. A highly selective separation is often a prerequisite for demonstrating specificity in the overall method.
The adjustment of chromatographic parameters—pH, column chemistry, and gradient profile—directly manipulates the physicochemical interactions that govern selectivity. By strategically optimizing these parameters, researchers can resolve critical peak pairs, such as separating a primary active pharmaceutical ingredient (API) from its degradation products, thereby providing the analytical specificity required for regulatory submissions [72] [7]. This guide details the systematic optimization of these parameters to achieve the precise balance between specificity and selectivity demanded by modern drug development.
Theoretical Foundation: The Resolution Equation and Its Parameters
Chromatographic optimization aims to maximize resolution (Rs), a measure of the separation between two adjacent peaks. Resolution is governed by the fundamental equation below, which breaks down into three key parameters: efficiency (N), retention factor (k), and selectivity (α) [73].
The Fundamental Resolution Equation:
Rs = (1/4) * √N * [(α - 1)/α] * [k₂/(1 + k₂)]
The following diagram illustrates how the primary chromatographic parameters influence these factors and, consequently, the final resolution of your separation.
Diagram 1: The relationship between chromatographic parameters and the factors of the resolution equation shows that selectivity (α) offers the most powerful leverage for improving separation.
Strategic Parameter Adjustment for Enhanced Separation
Mobile Phase pH Optimization
Mobile phase pH is a powerful tool for modulating selectivity, especially for ionizable analytes (weak acids or bases). A shift in pH alters the degree of ionization, changing the analyte's hydrophobicity and its interaction with the stationary phase [74].
- Mechanism of Action: For a basic analyte, lowering the pH below its pKa will protonate it, increasing its positive charge. In reversed-phase chromatography, this reduces retention as the charged species partitions more favorably into the aqueous mobile phase. The opposite effect occurs for acidic analytes [74].
- Optimization Strategy: A useful approach is to perform initial scouting runs with buffers at pH 3.0 and 7.0 to determine the sensitivity of the separation to pH changes. A common practice is to set the mobile phase pH at least 2 units away from the analyte's pKa to ensure it exists predominantly in one form, ensuring consistent and predictable retention [74].
- Impact on Specificity: Proper pH control is critical for separating analytes from impurities or degradants of similar structure but different pKa values. For instance, it can resolve an API from a degradant that has gained or lost an ionizable group.
Table 1: Effect of pH on Ionizable Analytes in Reversed-Phase HPLC
| Analyte Type | pKa Range | Recommended pH | Effect on Retention | Impact on Selectivity |
|---|---|---|---|---|
| Weak Acids | 3.0 - 5.0 | ≤ pKa - 2 (Protonated) | Increased retention (neutral form) | High for separating acids with small pKa differences |
| ≥ pKa + 2 (Deprotonated) | Decreased retention (anionic form) | |||
| Weak Bases | 5.0 - 8.0 | ≤ pKa - 2 (Protonated) | Decreased retention (cationic form) | High for separating bases with small pKa differences |
| ≥ pKa + 2 (Deprotonated) | Increased retention (neutral form) | |||
| Acid/Base Mixtures | Varies | Intermediate (e.g., 4.0-5.0) | Can maximize differences in ionization state | Very high, can dramatically alter elution order |
Chromatographic Column Selection
The stationary phase is the heart of the chromatographic separation. Its selection directly governs the thermodynamic interactions that define selectivity.
- Stationary Phase Chemistry: The primary mechanism in reversed-phase HPLC involves hydrophobic interactions. However, secondary interactions (e.g., hydrogen bonding, π-π interactions, ion-exchange) can be leveraged for difficult separations [73] [71].
- C18/Bonded Silica Phases: The most common phase, ideal for most neutral and non-polar compounds.
- Phenyl Phases: Provide π-π interactions with aromatic analytes, offering different selectivity for compounds with double bonds or aromatic rings.
- Polar-Embedded Phases: Contain polar groups (e.g., amide) that can improve peak shape for basic compounds and offer unique selectivity.
- Cyano Phases: A versatile phase with weak hydrophobicity and dipole interactions, suitable for both reversed-phase and normal-phase applications [74].
- Column Dimensions: Shorter columns (e.g., 50-100 mm) with smaller particle sizes (e.g., 1.7-3 µm) provide higher efficiency and faster separations, ideal for method development scouting. Longer columns (e.g., 150-250 mm) can be used to increase efficiency and resolution for complex mixtures [74].
Table 2: Guide to HPLC Stationary Phase Selection for Different Analyte Types
| Analyte Characteristics | Recommended Stationary Phase | Retention Mechanism | Application Example |
|---|---|---|---|
| Non-polar to medium polarity | C18, C8 | Hydrophobic | Paracetamol assay [72] |
| Aromatics, compounds with double bonds | Phenyl, Phenyl-Hexyl | Hydrophobic, π-π | Separation of structural isomers |
| Polar, basic compounds | Polar-embedded (e.g., amide), Cyano | Hydrophobic, H-bonding | Peptide analysis |
| Acidic and basic mixtures | Neutral C18 (high purity silica) | Hydrophobic, ion-suppression | Impurity profiling of ionizable APIs |
| Small, very polar molecules | HILIC, Cyano | Hydrophilic interaction, partitioning | Sugar analysis in nectar [75] |
Gradient Elution Optimization
Gradient elution, which involves changing the mobile phase composition over time, is essential for separating complex samples containing analytes with a wide range of hydrophobicity [74]. A key instrument parameter in gradient methods is the Gradient Delay Volume (GDV).
- Gradient Delay Volume (GDV): This is the volume between the point where solvents are mixed and the head of the column. The GDV causes a delay between the programmed gradient and its arrival at the column, impacting retention times and potentially selectivity [76].
- Optimization Strategy: The gradient profile (slope, time, and shape) can be optimized to balance resolution and analysis time. A steeper gradient reduces run times but may compromise resolution of critical pairs. A shallower gradient improves resolution but extends the analysis [72] [74]. For method transfer between instruments, the GDV must be accounted for, as differences can lead to failed separations [76].
- Impact on Specificity: A well-designed gradient ensures that all impurities and degradants are eluted and resolved from the main peak and from each other, which is a core requirement for demonstrating method specificity in impurity profiling [72] [7].
The workflow for developing and troubleshooting a gradient method is outlined below.
Diagram 2: A logical workflow for developing and optimizing a gradient elution method, incorporating iterative adjustments to the gradient profile and mobile/stationary phases to resolve critical pairs.
Advanced Optimization: The Design of Experiments (DoE) Approach
While the one-variable-at-a-time (OVAT) approach is common, it often fails to capture interactive effects between parameters. The Design of Experiments (DoE) methodology is a more efficient and powerful strategy for understanding complex systems [75].
A Box-Behnken Design (BBD), a type of Response Surface Methodology (RSM), allows for the simultaneous investigation of multiple factors (e.g., column temperature, acetonitrile concentration, flow rate) with a minimal number of experimental runs. The model evaluates both individual and interactive effects of these variables on critical responses like resolution between a critical peak pair [75].
- Case Study Application: In developing an HPLC method for sugars in sunflower nectar, a BBD was used to optimize three factors to resolve previously co-eluting peaks (glucose/mannitol and glucose/mannose). The model identified the optimal conditions (20°C, 82.5% acetonitrile, 0.766 mL/min), achieving a resolution (Rs) greater than 1 for all analytes [75].
- Benefits for Validation: The DoE approach provides a scientifically sound basis for establishing a Method Operable Design Region (MODR), demonstrating method robustness by showing that the method remains valid within a defined range of parameter variations [7].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for HPLC Method Development
| Reagent/Material | Function in Method Development | Application Example |
|---|---|---|
| Sodium Octanesulfonate | Ion-pairing reagent to modulate retention of ionizable analytes. | Determination of paracetamol and its impurity [72]. |
| Buffers (e.g., Phosphate, Acetate) | Control mobile phase pH to ensure stable ionization of analytes and reproducible retention. | Essential for separation of weak acids/bases; pH 3.2 used for paracetamol assay [72]. |
| HPLC-Grade Acetonitrile/Methanol | Organic modifier to control solvent strength and selectivity in reversed-phase HPLC. | Primary organic solvent in mobile phase; choice affects selectivity [74]. |
| Zorbax SB-Aq Column | Hydrophilic endcapped C18 column stable in aqueous mobile phases, good for polar analytes. | Separation of paracetamol, phenylephrine, and pheniramine [72]. |
| Nucleosil NH2 Column | Aminopropyl-bonded phase for polar compound analysis (e.g., sugars) via HILIC or normal phase. | Separation of sugars and sugar alcohols in nectar analysis [75]. |
| Uracil | Tracer compound with strong UV absorbance, used for measuring column dead time (t₀) and system GDV. | Experimental determination of Gradient Delay Volume [76]. |
Method Validation: Demonstrating Specificity and Robustness
Once a separation is optimized, its performance must be formally validated per International Council for Harmonisation (ICH) guidelines to confirm it is suitable for its intended purpose [7] [74].
- Specificity: The optimized method must demonstrate that the analyte peak is pure and free from interference from placebo, impurities, or degradants. This is typically shown by injecting samples containing all potential interferents and using a diode array detector to check peak purity [7].
- Robustness: This validation parameter evaluates the method's capacity to remain unaffected by small, deliberate variations in method parameters (e.g., flow rate ±0.1 mL/min, temperature ±2°C, pH ±0.1 units, mobile phase composition ±2%). A method developed using a DoE approach has built-in robustness data [7] [75].
- Linearity, Accuracy, and Precision: The method must demonstrate a linear response over the specified range, accuracy (recovery of 98-102% for APIs), and acceptable precision (repeatability and intermediate precision) [7].
The journey from a preliminary chromatographic method to a validated one is a systematic process of iterative optimization. By understanding the theoretical principles of separation, researchers can make intelligent adjustments to critical parameters—pH, column chemistry, and gradient profile—to engineer the selectivity necessary for a specific and robust analytical method.
Framing this work within the context of the broader thesis underscores a critical point: selectivity achieved through chromatographic separation is the practical foundation upon which the validation parameter of specificity is built. A method that cannot chromatographically resolve an API from its impurities cannot be considered specific, regardless of the detection technique. The strategies outlined herein, from fundamental parameter adjustment to advanced DoE workflows, provide a roadmap for developing reliable HPLC methods that meet the rigorous demands of pharmaceutical analysis and regulatory validation.
Assessing and Improving Peak Purity in Presence of Degradants and Impurities
In the realm of pharmaceutical analysis, the concepts of specificity and selectivity form the cornerstone of reliable analytical methods. While these terms are often used interchangeably, they carry distinct meanings: selectivity refers to a method's ability to measure the analyte accurately in the presence of potential interferents, whereas specificity represents the absolute ability to assess unequivocally the analyte in such a mixture [77]. Within this framework, peak purity assessment emerges as a critical technical procedure to demonstrate that a chromatographic peak represents a single chemical entity, thereby confirming the method's stability-indicating capability.
The pharmaceutical industry operates within a stringent regulatory landscape where International Conference on Harmonisation (ICH) guidelines mandate stress testing to identify likely degradation products, establish degradation pathways, and validate stability-indicating procedures [33]. Forced degradation studies are conducted under conditions more severe than accelerated stability testing to generate representative degradants, and peak purity assessment provides the necessary evidence that the analytical method can adequately resolve the active pharmaceutical ingredient (API) from these degradation products [78]. This technical guide explores the theoretical foundations, practical methodologies, and advanced techniques for accurate peak purity assessment within the context of analytical method validation.
Theoretical Foundations of Peak Purity Assessment
Fundamental Principles and Regulatory Expectations
Chromatographic peak purity verification is predicated on the fundamental principle that a pure compound will exhibit consistent spectral characteristics across all points of its elution profile. The presence of co-eluting compounds—whether impurities, degradants, or matrix components—manifests as detectable variations in these spectral properties [78]. The regulatory expectation, though not explicitly prescribed in method validation guidelines, has evolved such that peak purity assessment using photodiode array (PDA) detection has become a de facto standard for demonstrating method selectivity in regulatory submissions [78].
The ICH Q2(R1) guideline acknowledges that "peak purity tests may be useful to show that the analyte chromatographic peak is not attributable to more than one component," specifically mentioning diode array and mass spectrometry as potential techniques [78]. However, it stops short of mandating any specific approach, allowing flexibility based on scientific justification. This ambiguity necessitates that pharmaceutical companies develop robust, science-based strategies for peak purity assessment that satisfy regulatory expectations while maintaining technical soundness.
Critical Definitions: Specificity vs. Selectivity
In analytical chemistry, precise terminology is essential for clear communication and appropriate method characterization:
Specificity refers to the ability of a method to measure solely the analyte of interest without contribution from other components [77]. It represents an absolute concept—the method responds only to the target analyte.
Selectivity describes the ability of a method to quantify the analyte accurately despite the presence of other potentially interfering components [77]. Selectivity exists on a continuum, with methods being more or less selective toward specific interferents.
Quantitative approaches have been proposed to express selectivity and specificity as relative values ranging from 0 to 1, providing a numerical characterization of these method attributes [77]. For chromatographic methods, peak purity assessment serves as practical evidence of both specificity and selectivity by demonstrating that the target analyte peak is unaffected by co-eluting species.
Technical Approaches for Peak Purity Assessment
Photodiode Array (PDA) Detection
PDA-facilitated peak purity assessment represents the most widely employed technique in the pharmaceutical industry due to its accessibility, efficiency, and robust integration with liquid chromatography systems [79] [78]. The fundamental principle involves collecting full ultraviolet-visible spectra across the chromatographic peak—typically at the start, apex, and end positions—and comparing these spectra for homogeneity [79].
The technical implementation relies on sophisticated algorithms within chromatographic data systems (CDS) that perform the following sequence:
Spectral Collection: Continuous UV-Vis spectra are acquired throughout the elution of the chromatographic peak.
Baseline Correction: Spectra are corrected by subtracting interpolated baseline spectra between peak baseline liftoff and touchdown points.
Vector Transformation: Corrected spectra are converted into vectors in n-dimensional space, with vector lengths normalized using least-squares regression.
Spectral Contrast Calculation: The angle between spectral vectors is measured, with 0° indicating identical spectral shapes and 90° indicating no spectral overlap [78].
Commercial CDS platforms implement slightly different terminology and algorithms, though the core principles remain consistent:
Table 1: Peak Purity Algorithm Implementation in Commercial CDS Platforms
| Software Platform | Calculation Method | Purity Metric | Threshold Metric |
|---|---|---|---|
| Waters Empower | Spectral angle | Purity Angle | Purity Threshold |
| Agilent OpenLab | Similarity factor | 1000 × r² (where r = cosθ) | Reference spectrum comparison |
| Shimadzu LabSolutions | Cosine similarity | cosθ value | Built-in deconvolution (i-PDeA II) |
The fundamental decision rule states that a chromatographic peak is considered spectrally pure when the purity angle is less than the purity threshold [79]. The purity threshold incorporates uncertainty derived from spectral variation attributable to solvent and noise contributions, establishing the maximum allowable variation for a peak to be considered pure [79] [78].
Mass Spectrometry-Based Approaches
Mass spectrometry provides an orthogonal technique for peak purity assessment that complements PDA detection, particularly valuable when dealing with compounds having similar UV spectra or minimal chromophores [80] [78]. MS-based approaches detect co-eluting species through variations in mass-to-charge ratios rather than spectral characteristics.
The implementation typically involves:
Total Ion Chromatogram (TIC) Monitoring: Examining the TIC for unexpected peaks or shoulder formations.
Extracted Ion Chromatogram (EIC) Analysis: Comparing EICs for precursor ions, product ions, and adducts across different segments of the chromatographic peak.
Spectral Consistency Verification: Demonstrating consistent mass spectral profiles across the peak front, apex, and tail regions [78].
MS detection offers superior sensitivity for low-level impurities and can distinguish between isobaric compounds through fragmentation patterns. However, limitations include potential ionization suppression, differential ionization efficiencies between compounds, and higher instrumentation costs [78].
Orthogonal and Supplementary Techniques
Several supplementary approaches strengthen peak purity assessment when primary techniques yield ambiguous results:
Orthogonal Chromatographic Separation: Employing a second chromatographic method with different separation mechanisms (e.g., reversed-phase vs. hydrophilic interaction) to confirm resolution of potential co-eluters.
Two-Dimensional Liquid Chromatography (2D-LC): Comprehensive separation technology that subjects fractions from the first dimension to a second separation with different selectivity, providing exceptional resolution capability [78].
Spiking Studies: Introducing known impurities or degradation products into the sample to demonstrate adequate resolution from the main peak under method conditions.
Each technique offers distinct advantages and limitations, summarized in the following table:
Table 2: Comparison of Peak Purity Assessment Techniques
| Technique | Detection Principle | Key Advantages | Key Limitations |
|---|---|---|---|
| PDA Detection | UV Spectral homogeneity | Non-destructive; widely available; cost-effective | Limited for compounds with similar UV spectra; poor sensitivity for low-level impurities |
| Mass Spectrometry | Mass-to-charge ratio | High sensitivity; detects isobaric compounds; provides structural information | Ionization suppression; differential response factors; higher cost |
| 2D-LC | Orthogonal separation mechanisms | Superior separation power; comprehensive profiling | Method complexity; longer analysis times; potential solvent incompatibility |
| Spike Studies | Retention time matching | Confirms resolution of specific known compounds | Requires availability of impurity standards; limited to known compounds |
Implementing Forced Degradation Studies
Strategic Design and Conditions
Forced degradation studies represent a critical component of validating stability-indicating methods, intentionally generating degradants that might form during storage to demonstrate method capability [33]. A scientifically designed study incorporates multiple stress conditions while avoiding excessive degradation that produces irrelevant secondary degradants.
The general protocol includes the following stress conditions:
Table 3: Recommended Conditions for Forced Degradation Studies
| Degradation Type | Experimental Conditions | Typical Temperatures | Sampling Time Points |
|---|---|---|---|
| Acid Hydrolysis | 0.1 M HCl | 40°C, 60°C | 1, 3, 5 days |
| Base Hydrolysis | 0.1 M NaOH | 40°C, 60°C | 1, 3, 5 days |
| Oxidative Degradation | 3% H₂O₂ | 25°C, 60°C | 1, 3, 5 days |
| Thermal Degradation | Solid or solution state | 60°C, 80°C | 1, 3, 5 days |
| Photolytic Degradation | 1× and 3× ICH conditions | N/A | 1, 3, 5 days |
Reasonable degradation targets 5-20% for method validation, with 10% often considered optimal for small molecule pharmaceuticals [33]. Studies should be terminated if no degradation occurs after exposure to conditions exceeding accelerated stability protocols, as this indicates inherent molecule stability [33].
Sample Preparation and Concentration Considerations
Drug substance concentration typically begins at 1 mg/mL, which generally enables detection of minor degradation products [33]. Additional studies at expected formulation concentrations may be warranted, particularly for compounds prone to concentration-dependent degradation (e.g., aminopenicillins and aminocephalosporins) [33].
The Scientist's Toolkit: Essential Materials and Reagents
Successful implementation of peak purity assessment and forced degradation studies requires carefully selected materials and reagents. The following table catalogs essential components:
Table 4: Essential Research Reagent Solutions for Peak Purity Assessment
| Reagent/Material | Technical Function | Application Notes |
|---|---|---|
| High-Purity Water | Mobile phase component; sample preparation | LC-MS grade recommended to minimize background interference |
| Acid Solutions (HCl) | Forced degradation: acid hydrolysis | Typically 0.1-1.0 M concentrations; neutralization may be required before analysis |
| Base Solutions (NaOH) | Forced degradation: base hydrolysis | Typically 0.1-1.0 M concentrations; neutralization may be required before analysis |
| Hydrogen Peroxide | Forced degradation: oxidative stress | 1-3% concentrations; shorter exposure times (24h maximum) |
| Photodiode Array Detector | Spectral acquisition across UV-Vis range | Essential for PDA-based peak purity assessment |
| Mass Spectrometer | Mass-based detection and purity assessment | Single quadrupole sufficient for basic MS purity assessment |
| Chromatography Columns | Analytical separation | Multiple column chemistries recommended for orthogonal methods |
| Reference Standards | Method qualification and peak identification | Certified reference materials for API and available impurities |
Troubleshooting and Method Optimization
Addressing Common Challenges
False negative results (undetected co-elution) represent a significant risk in peak purity assessment and occur when co-eluting compounds exhibit minimal spectral differences, poor UV response, elution near the peak apex, or presence at very low concentrations [78]. Conversely, false positive results (pure peaks flagged as impure) may arise from significant baseline shifts due to mobile phase gradients, suboptimal data processing settings, interference from background noise, or measurements at extreme wavelengths (<210 nm or >800 nm) [78].
Mitigation strategies include:
Optimal Wavelength Selection: Choosing detection wavelengths with adequate analyte absorbance while avoiding extreme spectral regions prone to noise.
Appropriate Data Processing: Careful baseline placement, optimal integration parameters, and scientifically justified purity threshold settings.
Multi-Technology Correlation: Combining PDA results with mass balance calculations and orthogonal techniques to confirm findings.
Advanced Deconvolution Techniques
When conventional peak purity assessment suggests potential co-elution, advanced mathematical approaches can provide additional insight. The Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) algorithm, implemented in software such as Shimadzu's i-PDeA II, enables spectral deconvolution of partially resolved components [78]. These algorithms mathematically separate overlapping signals by iteratively refining pure component spectra and concentration profiles, potentially revealing impurities that conventional purity angle calculations might miss.
Peak purity assessment represents a critical element in demonstrating the specificity and selectivity of chromatographic methods, particularly for stability-indicating assays in pharmaceutical development. While PDA-based assessment serves as the industry standard, its limitations necessitate a holistic approach incorporating forced degradation studies, mass spectrometry, and orthogonal separations when appropriate. Through scientifically designed experiments and intelligent application of multiple assessment technologies, analysts can confidently verify method capability to accurately quantify APIs in the presence of degradants and impurities, ultimately ensuring drug product quality throughout its shelf life.
System Suitability Testing as a Tool for Ongoing Selectivity Assurance
In the framework of analytical method validation, selectivity is defined as the degree to which a method can quantify an analyte accurately in the presence of other target analytes or potential matrix interferences [12]. This distinguishes it from specificity, which traditionally refers to the ability to measure the analyte unequivocally in the presence of components that might be expected to be present, such as impurities, degradants, or matrix components [81]. While method validation provides initial evidence of a method's selectivity, this characteristic is not static and must be monitored throughout the method's operational lifecycle. System Suitability Testing (SST) serves as this critical ongoing assurance tool, verifying that the analytical system maintains the necessary selectivity each time it is used [82] [83].
SST functions as a real-time verification that the entire analytical system—comprising the instrument, reagents, column, and software—is performing within the predefined selectivity parameters established during validation [83]. For chromatographic methods, this means confirming that the system can adequately resolve the analyte of interest from potential interferents, ensuring that quantitation remains accurate and reliable for every analysis [84].
The Role of SST in Monitoring Chromatographic Selectivity
Key SST Parameters for Selectivity Assurance
System suitability tests for chromatographic methods evaluate several critical parameters that directly confirm the system's selective performance at the time of analysis [82].
Table 1: Key SST Parameters for Assessing Chromatographic Selectivity
| Parameter | Definition | Role in Selectivity Assurance | Typical Acceptance Criteria |
|---|---|---|---|
| Resolution (Rs) | Measures how well two adjacent peaks are separated, considering retention times and peak widths [82]. | Directly confirms baseline separation between analyte and closely eluting interferents [84]. | Typically >1.5 between critical pair [84]. |
| Tailing Factor (T) | Assesses peak symmetry, indicating possible active sites or secondary interactions [82]. | Ensures peak shape permits accurate integration and detection of partially co-eluting compounds [83]. | Typically <2.0 [84]. |
| Theoretical Plates (N) | Measures column efficiency under specific operating conditions [83]. | Indicates overall separation power of the chromatographic system [83]. | Method-specific minimum value. |
Establishing Selectivity Through SST Criteria
The SST criteria are established during method validation and must demonstrate that the method can withstand typical variations while maintaining selectivity [82]. The injection repeatability (precision), measured through the Relative Standard Deviation (RSD) of replicate injections of a standard, confirms the system's reproducibility, with the United States Pharmacopeia (USP) generally requiring an RSD of maximum 2.0% for five replicates [82]. The European Pharmacopoeia imposes even stricter requirements in some cases, with maximum repeatability as low as 1.27% for narrow specification limits [82]. The signal-to-noise ratio serves as a crucial SST parameter to ensure the method maintains sufficient sensitivity to detect and quantify analytes at the levels of interest, particularly for impurity methods [82].
Experimental Protocols for SST-Based Selectivity Verification
Preparation of System Suitability Test Solutions
The foundation of reliable SST is the proper preparation of test solutions. The sample and reference standard should be dissolved in the mobile phase or a comparable solvent to minimize baseline disturbances [82]. The concentration should be representative of the analytical range, typically at the level of quantitation for the analyte of interest. When dealing with complex matrices, a matrix blank and spiked solutions with known concentrations of analytes and potential interferents are essential for demonstrating selectivity during both validation and ongoing verification [12].
For impurity methods, it is critical to include a reference solution containing known impurities at specified levels to verify that resolution and sensitivity remain acceptable [85]. The FDA emphasizes that high-purity primary or secondary reference standards, qualified against former reference standards and not originating from the same batch as test samples, must be used for SST [82].
SST Execution and Acceptance Criteria Evaluation
The following workflow outlines the standard protocol for executing system suitability testing with a focus on selectivity verification:
For formal SSTs in pharmaceutical quality control, a minimum of five replicate injections of a standard are typically injected, with the calculated peak areas and chromatographic criteria objectively compared against predefined specifications [86]. The entire sequence—from system equilibration through final evaluation—must be documented to provide auditable evidence of system performance at the time of sample analysis.
Essential Research Reagent Solutions for Selectivity Assurance
The reliability of SST for ongoing selectivity monitoring depends on using appropriate, well-characterized reagents and materials throughout the analytical process.
Table 2: Essential Research Reagent Solutions for SST-Based Selectivity Assurance
| Reagent/Material | Function in Selectivity Assurance | Critical Quality Attributes |
|---|---|---|
| High-Purity Reference Standards | Serves as the performance benchmark for the system; verifies retention time stability, detector response, and peak shape [82]. | Certified purity, traceability to primary standards, stability under storage conditions. |
| Resolution Test Mixtures | Contains analytes and potential interferents to directly measure resolution between critical pairs [84]. | Stability, representative composition, coverage of expected interferents. |
| Matrix-Matched Blanks | Identifies potential matrix interferences that might co-elute with or affect quantification of the analyte [12]. | Representative matrix composition, consistency, absence of target analytes. |
| Column Efficiency Solutions | Contains compounds to measure theoretical plates and peak asymmetry under specific conditions [83]. | Stability, appropriate retention factor (k), well-characterized chromatographic behavior. |
| Mobile Phase Components | Creates the chromatographic environment that enables selective separation [82]. | HPLC-grade purity, low UV absorbance, minimal particulate matter. |
Regulatory Framework and Compliance Considerations
Regulatory authorities globally recognize the critical importance of SST for maintaining method selectivity throughout its operational life. The United States Pharmacopeia (USP) General Chapter <621> and the European Pharmacopoeia Chapter 2.2.46 provide specific guidance on SST requirements for chromatographic methods [82] [86]. Recent updates from the European Directorate for the Quality of Medicines & HealthCare (EDQM) have further clarified that when an assay references a related substances test procedure, the SST requirements from the purity test apply to the assay as well, reinforcing the integral role of SST in assuring selectivity [85].
The FDA explicitly states that if an assay fails system suitability, the entire run must be discarded, and no results should be reported other than the failure itself [82]. This regulatory position underscores the fundamental principle that analytical data generated on a system that has not demonstrated its suitability is inherently unreliable. Furthermore, regulators clearly distinguish between System Suitability Testing and Analytical Instrument Qualification (AIQ), emphasizing that SST is method-specific and does not replace the necessary qualification of the analytical instrument itself [82] [86].
Within the analytical method validation lifecycle, system suitability testing provides the essential bridge between initial validation data and daily operational assurance of method selectivity. By implementing robust, well-designed SST protocols that focus on critical separation parameters, laboratories can confidently verify that their analytical methods maintain the necessary selectivity to produce reliable results with each use. This ongoing verification is not merely a regulatory formality but represents a fundamental scientific practice that safeguards data integrity and ensures the quality and safety of pharmaceutical products.
Validation Protocols and Comparative Analysis: Ensuring Regulatory Compliance
Incorporating Specificity/Selectivity into Full, Partial, and Cross-Validation
Within the framework of analytical method validation, the concepts of specificity and selectivity represent foundational pillars for ensuring data quality, accuracy, and reliability. Although these terms are often used interchangeably, a subtle but crucial distinction exists, a nuance that has been formally clarified in modern regulatory guidelines such as ICH Q2(R2) [41]. Specificity refers to the ideal capability of a method to confirm the identity of a single analyte unequivocally, even in the presence of other components that may be expected to be present. It is the ability to assess the analyte without any ambiguity [1] [41]. In contrast, selectivity is the practical capability of a method to differentiate and quantify multiple analytes of interest from each other and from other components in the sample matrix, such as impurities, excipients, or degradation products [1] [41]. The relationship is hierarchical: a method that is specific is inherently selective, but a method can be selective without being specific for a single, unequivocal identity [41].
The proper demonstration of specificity and selectivity is not a one-time event but a continuous process that must be integrated into all stages of method validation, including full, partial, and cross-validation. As per the ICH M10 guideline, which establishes a harmonized global framework for bioanalytical method validation, the assessment of selectivity is now expected with greater rigor, requiring testing with multiple sources of biological matrix [87]. This technical guide explores how these core parameters are woven into the fabric of each validation type, providing drug development professionals with detailed protocols and data interpretation frameworks to ensure regulatory compliance and scientific integrity.
Core Concepts: Specificity and Selectivity
Definitions and Regulatory Context
A clear understanding of the definitions is the first step toward successful implementation. The following table summarizes the key differentiators between specificity and selectivity.
Table 1: Distinguishing Between Specificity and Selectivity
| Aspect | Specificity | Selectivity |
|---|---|---|
| Core Definition | The ability to assess unequivocally one analyte in the presence of potential interferents [1]. | The ability to differentiate and quantify multiple analytes from other components in the sample [41]. |
| Scope | Focused on a single analyte's identity [41]. | Encompasses the entire sample composition [1]. |
| Analogy | Using a unique key for a single lock [1]. | Identifying all keys in a keychain [1]. |
| ICH Q2(R2) Stance | Defined as a primary validation parameter [1] [41]. | Not directly defined, but noted as a demonstrable property when a method is not specific [41]. |
| Common Applications | Identification tests, assay of a single active ingredient [1]. | Related substances testing, impurity profiling, multi-analyte panels [41]. |
Experimental Assessment of Specificity and Selectivity
The experimental confirmation of specificity and selectivity follows a systematic approach designed to challenge the method with potential interferents.
For Specificity: The method is challenged by analyzing samples containing the analyte in the presence of other components, such as impurities, degradation products, or matrix components. Specificity is demonstrated when the response can be attributed solely to the analyte, with no interference from these other components. For chromatographic methods, this typically means the analyte peak is baseline-resolved from all other potential peaks [1].
For Selectivity: The method must be able to resolve and quantify all relevant analytes in the mixture. For a chromatographic method, this is demonstrated by the resolution of critical pairs of peaks. A common acceptance criterion is a resolution value (Rs) greater than 2.0 between any two adjacent peaks [41]. Selectivity assessments for bioanalytical methods, as per ICH M10, require testing matrices from at least six individual sources for chromatographic methods and ten for ligand-binding assays to account for biological variability [87].
The following diagram illustrates the logical workflow for assessing these parameters.
Integration into Full, Partial, and Cross-Validation
The extent and focus of specificity and selectivity testing vary significantly depending on the type of validation being performed. The following table summarizes the quantitative data and acceptance criteria for each validation type.
Table 2: Specificity/Selectivity Requirements Across Validation Types
| Validation Type | Objective | Minimum Selectivity Testing | Key Acceptance Criteria | Statistical Tools |
|---|---|---|---|---|
| Full Validation | Establish performance for a new method [87]. | 6 matrix sources for chromatography; 10 for LBA [87]. | No interference ≥20% of LLOQ for analyte/IS [87]. | Resolution factor (Rs > 2.0) [41]. |
| Partial Validation | Assess modified method [87]. | Test with new/modified interferents. | Comparable to original validated method. | As per the change (e.g., resolution). |
| Cross-Validation | Compare two validated methods [87]. | As per full validation, but for both methods. | Agreement between methods; No systematic bias. | Bland-Altman, Deming regression [87]. |
Full Validation
Full validation is conducted when a new bioanalytical method is established for the first time, typically for use in pivotal preclinical or clinical studies [87]. In this context, specificity and selectivity form the bedrock of the validation.
Experimental Protocol: A minimum of six independent sources of the appropriate biological matrix (e.g., human plasma) for chromatographic methods, and ten for ligand-binding assays, must be individually spiked with the analyte at the lower limit of quantitation (LLOQ) concentration and the internal standard (if used) [87]. These samples are then analyzed, and the responses are compared to those from blank matrices from the same sources. The guideline also recommends testing in lipemic and hemolyzed matrices when relevant to the patient population [87].
Acceptance Criteria: The mean analyte response in the LLOQ samples must meet predefined precision and accuracy criteria (typically ±20%). Most critically, in the corresponding blank samples, interference must be less than 20% of the LLOQ response for the analyte and less than 5% for the internal standard [87].
Partial Validation
Partial validation is performed when modifications are made to an already fully validated method. The scope of partial validation is determined by the nature of the change [87]. The integration of specificity and selectivity is targeted.
Scenarios Requiring Assessment:
- Change in Matrix: If switching from human plasma to urine, selectivity must be re-established in the new matrix.
- Change in Sample Processing: A new extraction procedure might introduce new reagents or plasticizers that could interfere, requiring a new selectivity challenge.
- Change in Instrumentation: A new detector (e.g., different mass spectrometer) might have different sensitivity to certain compounds, necessitating a check for new interfering peaks.
Experimental Protocol: The protocol is a subset of the full validation experiments, focusing on the areas impacted by the change. For instance, if a new anticoagulant is used in plasma collection, selectivity should be assessed using at least six lots of plasma containing the new anticoagulant.
Cross-Validation
Cross-validation is essential when data from two different bioanalytical methods, or from two different laboratories using the same method, are to be compared in a single study or program [87]. Its purpose is to ensure that the results are comparable and that there is no systematic bias between the methods.
Role of Specificity/Selectivity: Differences in the specificity or selectivity profiles of the two methods are a primary source of systematic bias. For example, one method might inadequately resolve a metabolite from the parent drug, while the other does not, leading to consistently different concentration readings.
Experimental Protocol: A common set of study samples, including incurred samples (samples from dosed subjects), are analyzed by both methods. The sample set should cover the entire calibration range and include QC samples.
Data Analysis and Statistical Tools: ICH M10 encourages the use of statistical techniques to evaluate agreement rather than rigid pass/fail criteria. Two recommended approaches are:
- Bland-Altman Analysis: Plots the difference between the two measurements against their average. This visualizes any systematic bias and its magnitude across the concentration range [87].
- Deming Regression: A type of linear regression that accounts for errors in both methods, providing a robust estimate of the slope and intercept, which indicate proportional and constant bias, respectively [87].
The following diagram outlines the cross-validation workflow with a focus on identifying bias stemming from specificity differences.
The Scientist's Toolkit: Essential Reagents and Materials
The successful execution of validation studies relies on a suite of critical reagents and materials. Proper characterization and documentation of these items are paramount, as emphasized by ICH M10, especially for large-molecule immunoassays [87].
Table 3: Key Research Reagent Solutions for Validation Studies
| Reagent/Material | Function | Critical Control Parameters |
|---|---|---|
| Reference Standard | Serves as the primary standard for quantifying the analyte; defines the calibration curve. | Identity, purity, certificate of analysis (CoA), storage conditions, and stability. |
| Internal Standard (IS) | Added to samples to correct for variability in sample processing and analysis; essential for LC-MS. | Stable isotope-labeling (e.g., ²H, ¹³C), purity, and absence of interference with the analyte. |
| Critical Reagents (LBAs) | Capture and detection antibodies, conjugated enzymes, or other binding molecules. | Specificity, affinity, lot-to-lot consistency, production method, storage, and stability [87]. |
| Biological Matrix | The material in which the analyte is quantified (e.g., plasma, serum, tissue homogenate). | Source (species), anticoagulant (for plasma), absence of inherent interference, and storage conditions. |
| Surrogate Matrix | Used for the quantification of endogenous compounds when a true blank matrix is unavailable. | Demonstrated equivalence to the natural matrix via parallelism testing [87]. |
Detailed Experimental Protocols
Protocol for Selectivity Assessment in a Bioanalytical Method
This protocol is designed to meet the requirements of ICH M10 for a chromatographic method (e.g., LC-MS) [87].
Materials Preparation:
- Obtain at least six individual lots of the relevant biological matrix (e.g., human plasma from six different donors).
- Prepare a stock solution of the analyte and the internal standard at a known, high concentration.
- Prepare an LLOQ working solution by serial dilution.
Sample Preparation:
- For each of the six matrix lots, prepare two sets of samples:
- Set A (LLOQ Samples): Spike the matrix with the analyte and IS at the LLOQ concentration (n=1 per lot).
- Set B (Blank Samples): Process the blank matrix with IS only (n=1 per lot). Also, process the blank matrix without IS to check for interference in the IS channel.
- For each of the six matrix lots, prepare two sets of samples:
Analysis:
- Analyze all samples (Set A and Set B from all six lots) in a single analytical run alongside a calibration curve.
Data Analysis and Acceptance Criteria:
- The mean calculated concentration for the LLOQ samples (Set A) must be within ±20% of the nominal value with a precision of ≤20% CV.
- For each blank sample with IS (Set B), the peak response in the analyte channel must be less than 20% of the mean peak response of the LLOQ samples.
- For each blank sample without IS, the peak response in the IS channel must be less than 5% of the mean IS response in the LLOQ samples.
Protocol for Cross-Validation Using Incurred Samples
This protocol is critical for bridging data between laboratories or methods [87].
Sample Selection:
- Select a minimum of 40 incurred samples (samples from subjects who have received the drug) from a previous study. These samples should cover the entire range of observed concentrations, including the Cmax (peak) and elimination phases.
Study Execution:
- Analyze the entire set of 40 samples using the original (reference) method (Method A) and the new/comparator method (Method B). The analysis order should be randomized to avoid sequence bias.
Data Analysis:
- For each sample, calculate the percent difference between the two methods:
% Difference = [(Method B - Method A) / Mean] * 100. - Perform Bland-Altman Analysis: Plot the % Difference for each sample against the mean concentration of Method A and Method B. Calculate the mean bias (average of all % differences) and the 95% limits of agreement (mean bias ± 1.96 * standard deviation of the differences).
- Perform Deming Regression: Plot the results from Method B against Method A. The ideal outcome is a slope of 1.0 and an intercept of 0.0.
- For each sample, calculate the percent difference between the two methods:
Acceptance Criteria:
- There should be no obvious systematic trend in the Bland-Altman plot.
- The 95% limits of agreement should be within pre-defined, clinically or analytically justified limits (e.g., ±30%).
- The 95% confidence interval for the slope from Deming regression should contain 1.0, and the 95% confidence interval for the intercept should contain 0.0. Any significant deviation indicates a proportional or constant bias, respectively, which must be investigated, potentially stemming from differences in method selectivity.
The incorporation of specificity and selectivity into the full lifecycle of bioanalytical method validation—from initial full validation through partial and cross-validation—is a critical determinant of data quality and regulatory success. The harmonized ICH M10 guideline provides a clear framework, elevating the expectations for selectivity testing, particularly through the use of multiple matrix lots and specialized matrices. Furthermore, its endorsement of sophisticated statistical tools like Bland-Altman analysis and Deming regression for cross-validation moves the field beyond simplistic pass/fail criteria and toward a more scientifically defensible, data-driven assessment of method comparability. By adhering to the detailed protocols and principles outlined in this guide, scientists can ensure their analytical methods are not only compliant but also robust, reliable, and capable of generating the high-quality data essential for informed decision-making in drug development.
Setting Acceptance Criteria for Resolution, Peak Purity, and Accuracy in Presence of Interferences
In analytical method validation, the concepts of specificity and selectivity are foundational to developing reliable methods. While the terms are often used interchangeably, a key distinction exists: selectivity refers to a method's ability to measure several analytes in a complex mixture, potentially with interference, whereas specificity is the ultimate degree of selectivity, indicating the method responds only to a single analyte [10]. This guide establishes the acceptance criteria for three critical parameters—resolution, peak purity, and accuracy in the presence of interferences—that empirically demonstrate a method's specificity. These validated criteria form the core of a robust control strategy, ensuring the reliability of data throughout the drug development lifecycle, from early development to commercial quality control, in compliance with modern regulatory guidelines like ICH Q2(R2) and ICH Q14 [21].
Regulatory and Scientific Framework
Regulatory bodies worldwide, including the FDA, EMA, and through the ICH, mandate rigorous specificity testing as part of method validation [88] [21]. The recent simultaneous issuance of ICH Q2(R2) and ICH Q14 signifies a shift from a prescriptive, "check-the-box" approach to a more scientific, risk-based, and lifecycle-based model [21]. Under this framework, the intended purpose of the analytical method should be defined prospectively in an Analytical Target Profile (ATP), which guides the development and validation process, including the setting of justified acceptance criteria [21].
The guidelines require demonstrating that an analytical procedure can unequivocally assess the analyte in the presence of potential interferents, such as impurities, degradation products, or matrix components [24] [21]. This is critical for avoiding false positives, inaccurate quantification, and ultimately, unreliable data that could compromise product quality and patient safety [88].
Core Acceptance Criteria and Their Experimental Determination
Resolution
Purpose: Resolution (Rs) quantitatively measures the separation between two adjacent chromatographic peaks. Sufficient resolution is critical for precise and rugged quantitative analysis, ensuring that the analyte peak is fully separated from any interfering peaks [10].
Acceptance Criterion: A resolution of Rs ≥ 2.0 between the analyte and the closest eluting potential interferent is generally required [88]. This value ensures baseline separation, which is crucial for accurate integration of both the main analyte and any nearby impurities.
Experimental Protocol:
- Prepare a System Suitability Solution: Create a mixture containing the analyte and all known available impurities at specified levels, typically at the level expected in the test samples or higher to challenge the method [24] [10].
- Chromatographic Analysis: Inject the solution and record the chromatogram.
- Calculate Resolution: Determine the resolution between the analyte and the most closely eluting impurity using the formula accepted by pharmacopoeias (e.g., USP). The calculation is typically automated by chromatography data systems.
Where
tis retention time andwis peak width.
Table 1: Summary of Acceptance Criteria for Specificity Parameters
| Parameter | Typical Acceptance Criterion | Critical For | Regulatory Reference |
|---|---|---|---|
| Resolution (Rs) | ≥ 2.0 | Peak separation, precise quantification | [88] |
| Peak Purity | Purity index / match threshold > 0.990 (or equivalent) | Confirming no co-elution | [88] |
| Accuracy in Presence of Interferences | Recovery within 98–102% (for assay) | Demonstrating lack of bias from interferents | [24] [89] |
Peak Purity
Purpose: Peak purity testing verifies that an analyte's chromatographic peak is attributable to a single component and is not obscured by a co-eluting substance. This is a direct test of a method's specificity [24] [88].
Acceptance Criterion: A purity index or match threshold greater than 0.990 is typically required when using a photodiode array (PDA) detector [88]. Some software systems may use a "pass/fail" result against a defined threshold.
Experimental Protocol:
- Utilize Advanced Detection: Perform analysis using a PDA detector or Mass Spectrometry (MS). PDA is common for LC-UV methods, while MS provides unequivocal confirmation [24].
- Collect Spectral Data: The PDA detector collects full UV spectra at multiple points across the peak (up-slope, apex, down-slope).
- Software Analysis: The instrument's software compares all the spectra within the peak. A pure peak will have highly similar spectra throughout. A changing spectrum within the peak indicates a potential co-elution.
- Forced Degradation Studies: To rigorously challenge peak purity, analyze stressed samples (e.g., exposed to acid, base, oxidant, heat, light) that have undergone approximately 5–20% degradation [88]. The peak purity of the main analyte must be demonstrated in these degraded samples.
Accuracy in the Presence of Interferences
Purpose: This parameter demonstrates that the accuracy (closeness to the true value) of the method is unaffected by the presence of impurities, degradation products, or matrix components [24] [21].
Acceptance Criterion: For a drug substance or product assay, accuracy is typically demonstrated by a recovery of 98–102% of the known, added amount for the analyte [24] [89]. This recovery must be met even in samples spiked with potential interferents.
Experimental Protocol:
- Sample Preparation: For a drug product assay, prepare a placebo blend (all excipients without the API). For impurity assays, use the drug substance or product.
- Spiking: Spike these samples with known quantities of the analyte and known quantities of all available impurities, degradants, or other potential interferents at levels expected or specified.
- Analysis and Calculation: Analyze the spiked samples and calculate the recovery of the analyte.
- Statistical Evaluation: The guidelines recommend data from a minimum of nine determinations over a minimum of three concentration levels (e.g., three concentrations, three replicates each) across the specified range [24]. The mean recovery and confidence intervals (e.g., ± standard deviation) should meet the acceptance criteria.
Table 2: Experimental Protocol for Key Specificity Tests
| Test | Recommended Samples to Analyze | Key Experimental Steps | Data Interpretation |
|---|---|---|---|
| Resolution | - System suitability mixture (analyte + impurities) [10] | 1. Prepare mixture2. Inject and run chromatogram3. Measure retention times and peak widths | Rs ≥ 2.0 between analyte and all nearest impurities |
| Peak Purity | - Standard solution- Stressed samples (5-20% degradation) [88]- Sample from stability studies | 1. Use PDA or MS detector2. Collect spectra across the peak3. Use software for purity assessment | Purity index > 0.990 confirms a homogeneous peak |
| Accuracy with Interference | - Placebo spiked with analyte & impurities [24]- Stressed samples | 1. Spike with known amounts2. Analyze multiple replicates3. Calculate % recovery | Mean recovery 98-102% demonstrates no bias from interferents |
Visualizing the Specificity Testing Workflow
The following diagram illustrates the logical workflow and decision points for establishing method specificity through the key experiments described in this guide.
Diagram 1: Specificity validation workflow and decision points.
The Scientist's Toolkit: Essential Reagents and Materials
A successful specificity study requires carefully selected and qualified materials. The following table lists key research reagent solutions and their critical functions in the validation process.
Table 3: Essential Research Reagent Solutions for Specificity Testing
| Reagent / Material | Function in Specificity Testing | Key Considerations |
|---|---|---|
| Highly Purified Analyte Reference Standard | Serves as the primary benchmark for identification, retention time, and spectral matching [24]. | Purity must be well-characterized; used to prepare calibration and system suitability solutions. |
| Authentic Impurity and Degradant Standards | Used to spike samples to challenge method selectivity and prove resolution from the main analyte [24] [10]. | Should include all known/suspected process impurities and forced degradation products. |
| Placebo Matrix (for Drug Product) | Represents the formulation without the active ingredient to test for interference from excipients [24]. | Must be representative of the final product composition. |
| Stressed Samples | Samples subjected to forced degradation (heat, light, pH, oxidation) to generate potential interferents and challenge peak purity [88]. | Aim for 5-20% degradation to create meaningful levels without destroying the analyte. |
| Appropriate Chromatographic Column | The stationary phase is a primary driver of selectivity; different chemistries (C18, phenyl, etc.) resolve compounds via different mechanisms [88]. | Select based on analyte properties; screening multiple columns may be necessary. |
| HPLC-Grade Solvents and Mobile Phase Additives | Form the mobile phase critical for achieving and maintaining separation and peak shape [88]. | Purity is essential to avoid ghost peaks and baseline noise that can interfere with detection. |
Setting scientifically sound and regulatory-justified acceptance criteria for resolution (Rs ≥ 2.0), peak purity (index > 0.990), and accuracy (recovery 98–102%) in the presence of interferences is non-negotiable for proving analytical method specificity. By integrating these criteria into a modern, lifecycle approach as defined in ICH Q2(R2) and Q14, and by employing a rigorous experimental workflow that includes forced degradation studies, scientists can develop robust, reliable methods. This comprehensive approach ensures the generation of high-quality data, safeguards product quality, and fulfills regulatory expectations from development through commercial control.
Within the framework of analytical method validation, specificity stands as a cornerstone parameter, ensuring that a method can accurately and unequivocally assess the analyte of interest in the presence of other potential components. The International Council for Harmonisation (ICH) defines specificity as the ability to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradants, and matrix components [24]. While often used interchangeably with selectivity, a crucial distinction exists: specificity represents the ideal state where an analyte is confirmed without any ambiguity, whereas selectivity refers to the practical capability to distinguish the analyte from other substances, often achieved with a resolution of >2 between interfering peaks [41]. In essence, a specific method is inherently selective, but a selective method may not be absolutely specific [41].
The demonstration of specificity is not a one-size-fits-all endeavor. The rigor and experimental focus required vary significantly depending on the analytical procedure's intended purpose. This paper provides a comparative analysis of how specificity requirements differ for three fundamental types of tests in pharmaceutical analysis: identification tests, assay tests, and impurity tests, framed within the broader context of specificity versus selectivity in analytical method validation research.
Specificity Fundamentals: Definitions and Regulatory Context
Specificity vs. Selectivity
The terms specificity and selectivity have often been blurred in analytical literature. Current guidelines, particularly ICH Q2(R2), have brought clarity. Selectivity is demonstrated when an analytical procedure can differentiate and quantify the analyte in the presence of other components like impurities, excipients, or degradation products. This is often practically assessed through chromatographic parameters such as resolution [41]. Specificity, on the other hand, is the ultimate ideal—the ability to confirm the identity of an analyte unequivocally, even in the presence of other components [41]. For example, a specific method would elute only the target analyte with no interference whatsoever.
The Regulatory Landscape
Analytical method validation, including the demonstration of specificity, is a regulatory requirement in the pharmaceutical industry to ensure the reliability and consistency of data. Key guidelines governing this area include:
- ICH Q2(R2): This is the primary international guideline for the validation of analytical procedures. It provides definitions and methodology for validation characteristics, including specificity/selectivity [90].
- FDA Guidance: The U.S. Food and Drug Administration aligns with ICH guidelines and provides specific expectations for method validation to support regulatory submissions [24] [90].
- USP (United States Pharmacopeia): The USP includes general chapters on method validation that are legally recognized in the United States [24].
A significant recent development is the update to ICH Q2(R2), which has streamlined validation requirements. A key change is the combined assessment of Specificity/Selectivity, emphasizing that tests must show an absence of interference from other substances and be specific to the target analyte [90]. Furthermore, the guidance now explicitly allows for the use of orthogonal methods to compensate for a lack of specificity in a single test [90].
Comparative Analysis of Specificity Requirements
The core objective of an analytical procedure dictates the nature and stringency of its specificity requirements. The following table provides a high-level comparison of these requirements across identification, assay, and impurity tests.
Table 1: Comparative Overview of Specificity Requirements
| Analytical Procedure | Primary Specificity Objective | Key Experimental Demonstrations | Critical Data Outputs |
|---|---|---|---|
| Identification Test | To confirm the identity of an analyte, not to quantify it [24]. | Comparison to known reference materials [91] [24]. Use of techniques that provide unique signatures (e.g., spectral matching) [24]. | Positive result for target analyte; negative result for similar, non-target compounds [89]. Visual or statistical match to reference (e.g., retention time, spectrum) [91]. |
| Assay Test | To quantify the major component(s) accurately [24]. | Resolution from closely eluting impurities/degradants [24]. Demonstration that the assay is unaffected by the presence of spiked impurities or excipients [24]. | Resolution between the main analyte and closest eluting potential interferent [24]. Peak purity confirmation via DAD or MS [24]. |
| Impurity Test | To detect and quantify minor components (impurities, degradants) alongside the major analyte [24]. | Resolution of all impurity peaks from each other and from the main analyte peak [91]. Forcing studies (stress testing) to generate degradants and demonstrate their separation [24]. | Impurity profile showing baseline separation of all components [91]. Peak purity for the main analyte to confirm no co-elution with impurities [24]. |
Specificity for Identification Tests
For identification tests, the central requirement for specificity is the ability to discriminate the target analyte from other closely related substances. The method must be capable of confirming "what" the substance is.
- Experimental Protocol: The primary methodology involves comparing the test sample's analytical response to that of a known reference standard [91] [24]. In chromatographic systems, this traditionally involves matching retention times with a reference standard. However, for higher specificity, techniques that provide a unique "fingerprint" are employed.
- Advanced Techniques: The use of peak purity tests based on photodiode-array (PAD or DAD) detection or mass spectrometry (MS) is highly recommended for unambiguous identification [24]. DAD detectors collect full spectra across a peak, and software algorithms compare these spectra to confirm the homogeneity of the peak. Mass spectrometry provides even greater specificity by confirming the identity based on molecular mass and fragmentation pattern [24].
- Acceptance Criteria: The test must yield positive results (correct identification) for samples containing the analyte and negative results for samples without it or with structurally similar compounds that could be mistaken for the analyte [89].
Specificity for Assay Tests
For assay procedures, which are used to quantify the major active component, specificity ensures that the measured response is solely due to the analyte of interest and that no other component interferes with the quantification.
- Experimental Protocol: Specificity is typically demonstrated by challenging the method with samples that contain potential interferents. This includes:
- Spiked Samples: The drug product (with excipients) is spiked with known impurities or degradants, and the assay's ability to quantify the active ingredient without interference is confirmed [24].
- Forced Degradation Studies: Also known as stress studies, these involve subjecting the sample to harsh conditions (e.g., acid, base, heat, light, oxidation) to generate degradants. The assay must then demonstrate that it can still accurately quantify the active ingredient in the presence of these degradation products [90].
- Chromatographic Parameters: The resolution (Rs) between the analyte peak and the closest eluting potential interferent (impurity, degradant, or excipient) is a critical value. A resolution of >2 is often considered indicative of a selective (and potentially specific) separation [41].
- Peak Purity Assessment: As with identification, the use of DAD or MS to demonstrate the homogeneity of the active pharmaceutical ingredient (API) peak is a powerful tool to prove specificity, showing that no other compounds are co-eluting with the main component [24].
Specificity for Impurity Tests
Impurity testing is arguably the most demanding application in terms of specificity requirements. The goal is not only to quantify the main component but also to resolve, detect, and accurately quantify often minute amounts of structurally similar impurities and degradants.
- Experimental Protocol: The core experiments involve the analysis of stressed samples and samples spiked with all available impurities [24]. The method must be able to elute and resolve all known and unknown impurities from each other and from the main peak to create a complete impurity profile [41] [91].
- The Challenge of "Unknowns": Since not all degradants may be known or available, a common practice is to compare the impurity profile obtained from the new method with that from a well-characterized orthogonal method [24]. Agreement between the two profiles supports the specificity of the new method.
- Peak Purity's Critical Role: Here, peak purity analysis is not just recommended but essential. It is used to confirm that the large main API peak is not hiding or co-eluting with a minor impurity, which could lead to an underestimation of impurity levels [24]. This is a situation where being "too specific" for the API alone is a disadvantage; the method must be selective enough to separate all components of interest [41].
The following workflow diagram illustrates the core experimental strategies employed to demonstrate specificity for each test type.
The Scientist's Toolkit: Essential Reagents and Materials
The experimental demonstration of specificity relies on a set of critical reagents and materials. The following table details these key items and their functions.
Table 2: Essential Research Reagents and Materials for Specificity Studies
| Reagent / Material | Function in Specificity Assessment |
|---|---|
| Highly Purified Reference Standard | Serves as the benchmark for confirming the identity and quantitative response of the analyte. Used in identification tests and for preparing calibration standards in assay and impurity methods [24] [89]. |
| Known Impurity Standards | Used to spike into samples to demonstrate that the method can resolve and quantify specific impurities in the presence of the main analyte and other components [24]. |
| Placebo/Excipient Mixture | A blend of all non-active ingredients in a drug product. Used to demonstrate the absence of interfering peaks from excipients at the retention times of the analyte and impurities [24]. |
| Forced Degradation Samples | Samples of the drug substance or product that have been intentionally stressed (e.g., with acid, base, peroxide, heat, light). These are used to generate potential degradants and challenge the method's ability to separate the analyte from degradation products [24] [90]. |
| Chemical Stress Agents | Reagents such as hydrochloric acid (HCl), sodium hydroxide (NaOH), hydrogen peroxide (H₂O₂), etc., used to create forced degradation samples [24]. |
| Chromatographic Columns | Columns of different chemistries (e.g., C8, C18, phenyl) are often evaluated during method development to achieve the necessary separation and selectivity for a specific test [24]. |
Advanced Considerations and Future Directions
The Role of Orthogonal Methods
In cases where a single analytical procedure lacks sufficient specificity, ICH guidelines acknowledge that a combination of two or more complementary procedures can be used to demonstrate overall specificity [90]. For example, denaturing gel electrophoresis might separate a protein monomer from a covalently linked dimer, but a secondary assay like size-exclusion chromatography may be needed to quantify non-covalently linked aggregates [90]. The use of hyphenated techniques like LC-DAD-MS is a powerful manifestation of this principle, providing simultaneous chromatographic separation, spectral purity, and mass confirmation.
Impact of ICH Q2(R2) Updates
The recent adoption of ICH Q2(R2) has refined the approach to specificity/selectivity. A key emphasis is that for techniques considered inherently specific (e.g., NMR, MS), additional experimental studies to demonstrate a lack of interference may not be required if scientifically justified [90]. This introduces a welcome element of flexibility and risk-based thinking into the validation process, focusing effort where it is most needed.
Specificity is a foundational but nuanced requirement in analytical method validation. Its demonstration is not uniform but is instead tailored to the critical objective of the analytical procedure. Identification tests demand high discrimination to confirm identity, often through spectral matching. Assay tests require a clear separation of the major analyte from potential interferents to ensure accurate quantification. Impurity tests present the greatest challenge, necessitating a method capable of resolving a complex mixture of chemically similar minor components from the major peak and from each other.
Understanding these distinctions is crucial for researchers and drug development professionals to design scientifically sound validation protocols that meet regulatory expectations. The evolving landscape, guided by ICH Q2(R2), encourages a pragmatic and risk-based approach, leveraging orthogonal techniques and modern technology like mass spectrometry to provide unequivocal evidence of a method's reliability. As analytical technologies continue to advance, the principles of specificity and selectivity will remain central to ensuring the quality, safety, and efficacy of pharmaceutical products.
Documentation and Regulatory Submission Strategies
In the pharmaceutical and medical device industries, regulatory submissions represent the definitive gateway to market access, legal compliance, and commercial success. These structured packages sent to authorities like the FDA or EMA demonstrate product safety, quality, and efficacy through comprehensive documentation [92]. Within this framework, the principles of specificity and selectivity from analytical method validation provide a powerful conceptual lens for designing submission strategies that withstand regulatory scrutiny. These concepts, when properly applied, ensure that submissions unequivocally demonstrate what regulators need to see while efficiently differentiating critical information from supporting data.
Specificity in analytical methodology refers to "the ability to assess unequivocally the analyte in the presence of components which may be expected to be present" [1]. Translated to regulatory strategy, this means designing documentation that precisely targets and demonstrates substantial equivalence or superiority without being derailed by extraneous information. Selectivity, conversely, describes "the ability to differentiate the analyte(s) of interest from endogenous components in the matrix or other components in the sample" [12] – or in regulatory terms, the capacity to address all relevant components of a submission while clearly differentiating their relative importance and relationships. This whitepaper explores how these methodological principles inform high-impact regulatory strategies across product development lifecycles.
Analytical Foundations: Specificity and Selectivity in Method Validation
Conceptual Definitions and Distinctions
In analytical chemistry, specificity and selectivity represent related but distinct methodological attributes crucial for validation. According to ICH Q2(R1) guidelines, specificity is "the ability to assess unequivocally the analyte in the presence of components which may be expected to be present" [1]. This concept can be visualized through a key analogy: identifying a single correct key from a bunch that opens a particular lock, without necessarily identifying all other keys in the bunch [1] [11].
Selectivity extends this concept further, requiring "the analytical method should be able to differentiate the analyte(s) of interest and internal standard from endogenous components in the matrix or other components in the sample" [1]. Using the same analogy, selectivity requires identification of all keys in the bunch, not just the one that opens the lock [11]. Fundamentally, specificity refers to a method's ability to respond to one single analyte, while selectivity applies when the method responds to several different analytes [1] [11].
Practical Implementation in Analytical Systems
Method specificity is demonstrated through testing for interference in the presence of potentially confounding substances like impurities, degradation products, or matrix components [1]. For chromatographic methods, specificity is demonstrated by resolution between closely eluting peaks [1]. System suitability testing ensures system performance both before and after testing unknowns, verifying resolution and reproducibility [12].
Selectivity is established by demonstrating that a method can accurately quantify an analyte alongside other target analytes or matrix interferences [12]. While studying every possible interference is impractical, researchers should identify and test against the most likely and worst interferences [12]. Practical tools for establishing both parameters include blank samples (reagent and matrix blanks) and spiked solutions with known analyte concentrations to measure recovery and assess interference [12].
Table 1: Key Validation Parameters in Analytical Method Development
| Parameter | Definition | Experimental Approach | Acceptance Criteria |
|---|---|---|---|
| Specificity | Ability to measure analyte accurately despite interferences | Compare analyte response in presence/absence of potential interferents; forced degradation studies | No interference observed; peak purity demonstrated |
| Selectivity | Ability to differentiate and quantify multiple analytes in complex matrices | Resolve all analytes of interest; demonstrate separation from matrix components | Resolution factor ≥1.5 between critical pairs |
| Linearity | Results proportional to analyte concentration | Minimum 5 concentration points across specified range | Correlation coefficient ≥0.99 |
| LOD | Lowest concentration reliably detected | Signal/noise ratio approach | Typically 3×signal/noise |
| LOQ | Lowest concentration reliably quantified | Signal/noise ratio approach | Typically 10×signal/noise with precision ≤20% RSD |
Regulatory Submission Framework: Principles and Pathways
Submission Types and Strategic Considerations
Regulatory submissions follow distinct pathways based on product type and development stage. Each pathway requires specific documentation strategies aligned with its unique evidentiary requirements [92].
Table 2: Key Regulatory Submission Pathways and Requirements
| Submission Type | Purpose | Authority | Key Documentation Elements |
|---|---|---|---|
| IND | Initiate clinical trials | FDA | Preclinical data, manufacturing information, clinical protocols |
| NDA | New drug approval | FDA | Complete clinical evidence, CMC, labeling, safety data |
| ANDA | Generic drug approval | FDA | Bioequivalence data, CMC, reference product comparison |
| BLA | Biologics approval | FDA | Comprehensive clinical data, manufacturing controls |
| 510(k) | Device clearance | FDA | Substantial equivalence demonstration, performance testing |
| PMA | Device approval | FDA | Clinical safety effectiveness data, manufacturing information |
| MAA | Marketing authorization | EMA | Comprehensive quality, safety, efficacy data per EU requirements |
The Substantial Equivalence Principle
For medical devices following the 510(k) pathway, substantial equivalence represents a specificity challenge – demonstrating that a new device is sufficiently similar to a legally marketed predicate in both intended use and technological characteristics [93]. This requires strategic predicate device selection based on thorough understanding of intended claims and competitive landscape [93]. When significant technological differences exist, reference devices can strengthen submissions by providing additional comparison points [93].
Strategic Integration: Applying Specificity and Selectivity to Submission Design
Specificity in Regulatory Strategy
Strategic specificity begins with "simplification of filing strategy" built on rigorously defining and targeting the desired product label [94]. This promotes cross-functional collaboration between biostatistics, clinical development, regulatory, and safety teams, ensuring study designs meet requirements while focusing on critical-path activities [94]. Clinical programs designed by regulatory strategists with laser focus on efficiently demonstrating a product's benefit-risk profile combine with proactive health authority engagement to create precisely targeted submissions.
The pre-submission process offers invaluable opportunity to receive FDA feedback before committing to full submission strategy [93]. This specificity-enhancing step is particularly crucial when unsure about predicate device selection, when devices have significant differences from chosen predicates, or when testing plans deviate from established guidance [93]. Early interaction with reviewers identifies potential issues before they become roadblocks and aligns expectations, potentially streamlining formal review.
Selectivity in Comprehensive Documentation
Selectivity in regulatory submissions manifests through "zero-based redesign of submission process" that fundamentally rethinks documentation from the last patient's last visit through filing [94]. This involves strategic selection and parallel processing of multiple components:
- Data preparation optimization: Rigorous data collection, automated cleaning, and pre-arranged sign-offs [94]
- Predrafting of lean clinical reports: Early alignment on key messages, scenario-based front-loading, and application of lean writing principles [94]
- Batch generation and TLF standardization: Early alignment on key data, fixed TLF datasets, pre-programming, and standardized templates [94]
- Strategic review in 24 hours: Fixed document review times, restricted access to sections, and condensed reviewer matrix [94]
This selective approach ensures comprehensive coverage while differentiating critical from supplementary information, much like analytical selectivity distinguishes multiple analytes in complex mixtures.
Experimental Protocols and Methodologies
Method Validation Protocol for Specificity and Selectivity
Purpose: To establish and validate the specificity and selectivity of an analytical method for drug substance quantification in presence of potential interferents.
Materials and Reagents:
- Reference standards: Drug substance, known impurities, degradation products
- Matrix blanks: Placebo formulation containing all excipients
- Forced degradation samples: Acid/base hydrolysis, oxidative, thermal, photolytic stress conditions
- Chromatographic system: HPLC/UPLC with PDA or MS detection
Procedure:
- Prepare individual solutions of drug substance and each potential interferent at expected concentration levels
- Prepare mixture solutions containing drug substance and all potential interferents
- Inject placebo formulation to identify excipient interference
- Inject forced degradation samples to demonstrate separation of degradation products
- Analyze all samples using the proposed chromatographic method
- Assess peak purity using PDA or MS detection
- Calculate resolution between closest eluting peaks
Acceptance Criteria:
- Placebo injection shows no interference at retention time of analyte
- Peak purity index ≥0.99 for drug substance peak in all samples
- Resolution ≥2.0 between drug substance and closest eluting potential interferent
- No co-elution of any significant peak in mixture samples
Substantial Equivalence Testing Protocol
Purpose: To demonstrate substantial equivalence between a new medical device and predicate device through performance testing.
Materials:
- Test devices: Minimum of 3 lots of new device
- Control devices: Predicate device and/or reference devices
- Testing equipment: Calibrated according to manufacturer specifications
- Test samples: Appropriate biological or synthetic matrices
Procedure:
- Define critical performance parameters based on intended use and technological characteristics
- Establish testing protocols for each parameter following recognized standards
- Conduct side-by-side testing of new device and predicate/reference devices
- Perform statistical analysis comparing performance data
- Document all testing procedures, results, and analysis methods
Acceptance Criteria:
- New device performance within predetermined equivalence margins
- No statistically significant inferiority in safety or effectiveness
- All performance parameters meet or exceed predicate device specifications
Visualization of Strategic Approaches
Strategic Approaches to Regulatory Documentation
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Analytical Validation
| Reagent/Material | Function | Application in Validation |
|---|---|---|
| Reference Standards | Certified materials of known purity and identity | Quantification, method calibration, system suitability |
| Matrix Blanks | Sample matrix without analyte | Specificity testing, interference assessment |
| Forced Degradation Samples | Stressed samples containing degradation products | Specificity demonstration, stability-indicating method validation |
| Spiked Solutions | Samples with known added analyte concentrations | Recovery studies, accuracy determination, selectivity assessment |
| System Suitability Test Mixtures | Reference mixtures of critical analytes | Daily verification of chromatographic performance |
| Placebo Formulations | Complete formulation without active ingredient | Interference testing for assay and impurity methods |
Technological Enablers: AI and Automation in Modern Submissions
Technology plays an increasingly crucial role in achieving both specificity and selectivity in regulatory submissions. Modern, integrated core systems like regulatory-information-management systems (RIMS) enable seamless workflows, embedded automation, and data-centric approaches that replace document-heavy processes [94]. Automation specifically targets time-consuming formatting of tables, listings, and figures – currently automated at scale by only 13% of companies – offering substantial efficiency gains [94].
Generative AI represents a transformative technology for enhancing regulatory selectivity. Early pilots demonstrate that gen-AI-assisted medical writing can reduce end-to-end cycling time for clinical-study report authoring by 40% [94]. One AI-powered platform reduced first-draft writing time from 180 hours to 80 hours while cutting errors by 50% [94]. These technologies enable more selective attention to critical content while automating routine documentation tasks.
Technology-Enabled Submission Workflow
The principles of specificity and selectivity from analytical method validation provide a robust framework for designing effective regulatory submission strategies. Specificity ensures targeted, unequivocal demonstration of key claims, while selectivity enables comprehensive documentation that differentiates multiple evidentiary elements. Together, these principles guide development of submissions that successfully navigate regulatory review, accelerating patient access to innovative therapies while maintaining rigorous safety and efficacy standards. As regulatory landscapes evolve, the integration of these methodological principles with advanced technologies like AI and automation will further transform submission excellence across the pharmaceutical and medical device industries.
In the rigorous world of pharmaceutical development, demonstrating the specificity and selectivity of analytical methods is a fundamental regulatory requirement that remains a persistent challenge. These two parameters form the bedrock of reliable data, ensuring that a method can accurately and unequivocally measure the analyte of interest in the presence of other components. Despite their established importance, deficiencies in adequately proving specificity and selectivity consistently rank among the most frequent audit findings by regulatory agencies globally [95] [96].
The International Council for Harmonisation (ICH) provides the foundational definitions that frame this discussion. In the context of analytical procedures, specificity is the ability to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradants, or matrix components [97]. It is often considered the ultimate proof of a method's reliability. Selectivity, while sometimes used interchangeably, refers to a procedure's ability to measure a particular analyte in a mixture without interference from other analytes in that mixture. It can be viewed as a spectrum of discrimination, whereas specificity implies absolute exclusivity [98]. The recent adoption of ICH Q14 on analytical procedure development and the revision of ICH Q2(R2) underscore the heightened regulatory focus on a more structured, science-based approach to developing and validating these critical method attributes [99] [97]. A failure to adequately address them not only triggers audit observations but can compromise product quality and patient safety.
Regulatory Frameworks and the Impetus for Audit Findings
The regulatory landscape for analytical procedures has evolved significantly, moving from a checklist approach to an integrated lifecycle concept. The finalization of ICH Q14 and Q2(R2) in 2023-2024 marks a significant shift, expecting a more profound understanding of method performance and its control [99] [97]. Under this enhanced framework, regulatory evaluations now scrutinize the scientific rationale behind method development, demanding robust risk assessment and a clearly defined Analytical Target Profile (ATP) [99]. The ATP is a prospective summary of the required quality characteristics of an analytical procedure, defining its purpose and the performance criteria it must meet throughout its lifecycle. A poorly constructed ATP that does not adequately define specificity and selectivity requirements is a common root cause of later audit findings.
Audit findings related to specificity and selectivity often stem from a disconnect between the traditional, linear approach to method validation and the modern, holistic Analytical Procedure Lifecycle Management (APLM) concept. As one industry expert notes, "Using the tools described in the ICH guidance for industry Q12... the guidance describes principles to support change management of analytical procedures based on risk management, comprehensive understanding of the analytical procedure, and adherence to predefined criteria for performance characteristics" [97]. Findings frequently cite a lack of "analytical robustness," where methods, while seemingly specific under ideal conditions, fail when subjected to minor, but realistic, variations in parameters [96]. This indicates an insufficient investigation of the Method Operable Design Region (MODR) during development, a key expectation of the enhanced approach in ICH Q14 [99].
Common Pitfalls Identified in Audit Findings
An analysis of recurring audit observations reveals a pattern of specific deficiencies in demonstrating specificity and selectivity. These pitfalls can be broadly categorized into strategic, experimental, and documentation failures.
Inadequate Forced Degradation Studies
One of the most critical audit findings involves incomplete or poorly designed forced degradation studies intended to demonstrate the stability-indicating power of a method. Common shortcomings include:
- Insufficient Degradation: Failing to achieve the recommended 5-20% degradation of the active ingredient, which prevents meaningful assessment of peak purity and separation.
- Non-Representative Conditions: Using stress conditions (e.g., extreme pH, temperature) that are not relevant to the drug substance's or product's actual stability profile.
- Missing Intermediates: A failure to identify and monitor key degradation intermediates, which could co-elute with the main peak under routine stability conditions.
- Poor Chromatographic Resolution: Not optimizing the method to achieve baseline separation between the analyte and all known degradants, relying instead on peak purity algorithms alone, which can be misleading with co-eluting compounds of similar spectra [95].
Failure to Assess Matrix Interference
For bioanalytical or impurity methods, a frequent finding is the inadequate characterization of matrix effects. This is a paramount selectivity challenge. Pitfalls include:
- Testing Inadequate Matrix Lots: Using too few lots of blank matrix (e.g., plasma, serum, or placebo formulation) to account for natural biological or compositional variation.
- Ignoring Matrix Components: Failing to investigate the potential for interference from metabolites, excipients, or concomitant medications, leading to inaccurate quantification [95] [96].
- Incomplete Evaluation of Sample Preparation: Not demonstrating that the sample cleanup procedure consistently and effectively removes interfering components across different matrix lots.
Over-reliance on a Single Detection Technique
Audits often identify a lack of orthogonal method support for claims of specificity. A common but deficient practice is to rely solely on chromatographic retention time in HPLC-UV as proof of identity and purity. Regulatory guidance expects, particularly for complex molecules like biologics, that additional techniques such as mass spectrometry (MS) or photodiode array (PDA) detection be used to confirm peak homogeneity and identity [96]. As one expert points out, "For new types of molecules and/or conjugate products... A direct potency test method may not exist, and instead, several surrogate test methods may need to be used" [96], highlighting the need for a multi-pronged approach to demonstrate specificity for novel modalities.
Poorly Defined and Controlled Critical Method Parameters
A fundamental pitfall is the failure to identify, understand, and control the Critical Method Parameters (CMPs) that govern specificity and selectivity. This often manifests as:
- Inadequate Robustness Testing: Not using structured approaches like Design of Experiments (DoE) to systematically evaluate how variations in parameters (e.g., mobile phase pH, column temperature, gradient slope) impact resolution and selectivity [99].
- Narrowly Defined Set Points: Establishing method conditions at a single, inflexible set point without understanding the Proven Acceptable Ranges (PAR). This makes the method vulnerable to failure with minor, routine equipment or reagent variations, a frequent audit observation [99].
Table 1: Summary of Common Audit Pitfalls and Their Technical Root Causes
| Audit Finding | Technical Root Cause | Potential Impact on Data |
|---|---|---|
| Incomplete Forced Degradation Study | Lack of relevant degradation pathways explored; insufficient degradation achieved. | Inability to detect key degradants during stability studies, risking patient safety. |
| Inadequate Matrix Assessment | Use of too few matrix lots; failure to test for interference from metabolites/excipients. | Inaccurate potency or impurity results due to undetected signal suppression or enhancement. |
| Over-reliance on Single Technique | Lack of orthogonal method (e.g., LC-MS) to confirm chromatographic purity. | False positives/negatives for impurities; misidentification of analytes. |
| Poor Robustness Testing | Failure to use DoE to understand impact of parameter variation on selectivity. | Method failure during transfer or routine use, leading to out-of-specification (OOS) results. |
| Uncontrolled Method Parameters | CMPs not identified; no established PAR or MODR for critical parameters affecting selectivity. | Lack of method reliability and reproducibility, triggering regulatory scrutiny. |
Proven Experimental Protocols to Overcome Pitfalls
Addressing common audit findings requires implementing rigorous, well-documented experimental protocols.
A Systematic Protocol for Forced Degradation Studies
A robust forced degradation study should be systematic and comprehensive.
- Objective: To demonstrate the stability-indicating capability of the method by intentionally degrading the sample and proving the method can separate degradants from the analyte.
- Materials: Drug substance, drug product, relevant solvents, and reagents for stress conditions.
- Stress Conditions:
- Acidic/Basic Hydrolysis: Use 0.1-1M HCl or NaOH at ambient and elevated temperatures (e.g., 40-60°C) for several hours.
- Oxidative Stress: Expose to 0.1-3% hydrogen peroxide at ambient temperature for several hours or days.
- Thermal Stress: Solid and solution state at elevated temperatures (e.g., 70-80°C).
- Photolytic Stress: Expose to controlled UV and visible light as per ICH Q1B.
- Procedure: For each condition, prepare samples in duplicate and analyze alongside unstressed and placebo samples (for drug product). Target 5-20% degradation.
- Analysis: Employ the primary method (e.g., HPLC-UV) and an orthogonal technique (e.g., HPLC-MS). Assess peak purity using a PDA detector. Ensure baseline separation of all degradant peaks from the main peak and from each other.
- Documentation: Report degradation profiles, peak purity results, and mass balance calculations.
A Comprehensive Protocol for Assessing Selectivity and Matrix Interference
This protocol is critical for bioanalytical methods or methods analyzing complex formulations.
- Objective: To prove the method can distinguish the analyte from other components in the sample matrix.
- Materials: At least six independent lots of blank matrix (e.g., plasma from different donors, placebo formulation from different batches).
- Procedure:
- Analyze each blank matrix lot.
- Analyze each blank matrix lot spiked with the analyte at the Lower Limit of Quantification (LLOQ).
- Analyze each blank matrix lot spiked with known or potential interferents (metabolites, excipients, concomitant drugs) at expected high concentrations.
- For impurity methods, analyze samples spiked with impurities at the specification level.
- Acceptance Criteria: Interference at the retention time of the analyte in blank samples should be <20% of the LLOQ response. The response for the LLOQ samples should have precision and accuracy within ±20%.
- Documentation: Provide chromatographic overlays of all blank and spiked matrices, demonstrating the absence of interference.
Diagram 1: Selectivity Assessment Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Successfully navigating specificity and selectivity challenges requires the use of well-characterized reagents and materials. The following table details key items essential for conducting the experiments described in this guide.
Table 2: Key Research Reagent Solutions for Specificity and Selectivity Studies
| Reagent / Material | Function in Specificity/Selectivity Studies | Critical Quality Attributes |
|---|---|---|
| High-Purity Reference Standards | To identify the analyte's retention time and spectral characteristics; used as a benchmark for peak purity and identification. | Certified purity (>98.5%), well-documented structure and chromatographic behavior, stability under storage conditions. |
| Forced Degradation Reagents | To intentionally generate degradants (e.g., HCl/NaOH for hydrolysis, H₂O₂ for oxidation) for stability-indicating method validation. | Defined concentration and purity, absence of interfering impurities, stability over the study duration. |
| Blank Matrix Lots | To assess and rule out matrix interference in bioanalytical or complex formulation analysis. | Representative of the test population (e.g., human plasma from ≥6 donors), well-documented source and handling, free of the analyte and interferents. |
| Known Impurity Standards | To confirm the method can separate and accurately quantify specified impurities and potential degradants. | Certified identity and purity, availability at required concentration levels. |
| Chromatographic Columns | The primary tool for achieving separation; different selectivities are often needed for optimal resolution. | Multiple column chemistries (C18, phenyl, HILIC, etc.), consistent batch-to-batch performance, and high chromatographic efficiency. |
Implementing a Risk-Based Lifecycle Approach
The most effective strategy to prevent audit findings is to adopt the proactive, knowledge-driven philosophy embedded in the latest ICH guidelines. ICH Q14 encourages an "enhanced approach" where understanding and controlling variability throughout the method's lifecycle is paramount [99]. This begins with a well-defined ATP that explicitly states the required specificity and selectivity, guiding all subsequent development and validation activities.
A core component of this approach is the implementation of a formal Analytical Control Strategy. This strategy involves identifying potential sources of variability—whether from the system, user, or environment—and implementing controls to mitigate their impact [99]. For specificity and selectivity, this means:
- Defining system suitability tests (SSTs) that directly monitor the critical separation (e.g., resolution between a critical pair of peaks) to be checked before every analytical run.
- Establishing Established Conditions (ECs) for the method parameters that most profoundly affect selectivity. As noted in the challenges of implementing ICH Q14, "Because reported ECs are considered legally binding, any changes to them must be justified and evaluated based on their risk categorizations" [99]. Understanding which parameters are high-risk allows for more focused control and easier, more flexible post-approval changes.
Diagram 2: Lifecycle Approach to Specificity & Selectivity
Finally, continuous monitoring of method performance during routine use is vital. Trends in SST data, such as a gradual decrease in resolution for a critical peak pair, can provide an early warning of a developing selectivity issue, allowing for proactive remediation before an analytical failure or audit finding occurs [99]. This shift from a one-time validation event to a holistic lifecycle management approach is the most powerful defense against common pitfalls in demonstrating specificity and selectivity.
Best Practices for Method Transfer and Inter-laboratory Ruggedness
Within the context of analytical method validation, the concepts of specificity and selectivity establish a method's fundamental ability to measure the analyte accurately in the presence of potential interferants [100]. Specificity is the gold standard, proving that a method can unequivocally assess the analyte in the presence of components like impurities, degradants, or matrix elements [32] [100]. Selectivity, often used interchangeably but implying a gradation, describes the method's ability to distinguish the analyte from a limited number of other components.
This foundational requirement directly influences the next critical challenge: ensuring the method's reliability when transferred between laboratories, instruments, and analysts. This reliability is encapsulated by the concept of ruggedness (also referred to as robustness), which is the measure of a method's capacity to remain unaffected by small, deliberate variations in procedural parameters [100]. In essence, while specificity confirms the method works under ideal, controlled conditions, ruggedness demonstrates that this performance is maintained in the real world. A method's inter-laboratory ruggedness is the ultimate expression of its robustness and a critical determinant for a successful technology transfer [101]. It validates that the method's specificity is not a fragile property of a single laboratory's environment but is reproducible and dependable across the global development and quality control network.
Key Approaches to Analytical Method Transfer
Selecting the appropriate transfer strategy is a critical decision that should be based on the method's complexity, its validated status, the experience of the receiving laboratory, and the associated risk [102] [103]. Regulatory bodies like the USP (General Chapter <1224>) provide guidance on several accepted approaches [102] [104].
Table 1: Comparison of Analytical Method Transfer Approaches
| Transfer Approach | Core Principle | Best Suited For | Key Considerations |
|---|---|---|---|
| Comparative Testing [102] [103] [105] | Both laboratories (Transferring and Receiving) analyze the same set of homogeneous samples. Results are statistically compared against pre-defined acceptance criteria. | Well-established, validated methods; laboratories with similar capabilities. | Requires careful sample preparation and homogeneity; robust statistical analysis plan is essential. |
| Co-validation [102] [106] [101] | The analytical method is validated simultaneously by both the transferring and receiving laboratories as part of a single, collaborative study. | New methods being developed for multi-site use from the outset. | High level of collaboration and harmonization required; data is presented in a single validation package. |
| Revalidation [102] [101] [105] | The receiving laboratory performs a full or partial revalidation of the method, treating it as new to their site. | Significant differences in lab conditions/equipment; substantial method changes; when the transferring lab is unavailable. | Most resource-intensive approach; requires a full validation protocol and report. |
| Transfer Waiver [102] [105] | The formal transfer process is waived based on strong scientific justification and documented risk assessment. | Highly experienced receiving lab using identical conditions; simple, robust methods (e.g., pharmacopoeial methods). | Carries high regulatory scrutiny; requires extensive documentation and QA approval. |
Detailed Protocol: Comparative Testing
Comparative testing is the most frequently used transfer approach [103] [105]. The following protocol outlines the key steps:
- Experimental Design: A minimum of one batch for an Active Pharmaceutical Ingredient (API) or one batch each for the lowest and highest strength of a drug product is typically used [104]. For a robust study, a minimum of six independent test setups (e.g., two analysts from the Receiving Laboratory (RL) each performing three replicates) is recommended to effectively assess precision [107].
- Sample Preparation: Homogeneous samples from the same lot are distributed to both the Transferring Laboratory (TL) and RL [102] [103]. Samples can include approved drug product, expired batches (if justified), or spiked samples where impurities are added to a placebo or active material to challenge method accuracy [103] [105].
- Execution: Both laboratories analyze the samples following the identical, approved analytical procedure. The RL should have completed all necessary training and equipment qualification prior to testing [103].
- Data Analysis and Acceptance Criteria: Results from both labs are statistically compared using pre-defined methods. Common acceptance criteria include [105]:
- Assay: Absolute difference between the mean results from TL and RL should typically not exceed 2-3%.
- Related Substances (Impurities): For impurities present at low levels (e.g., below 0.5%), recovery criteria of 80-120% for spiked impurities are common. For higher-level impurities, absolute difference criteria may be applied.
- Dissolution: Absolute difference in mean results is typically no more than 10% at time points when less than 85% is dissolved, and no more than 5% when more than 85% is dissolved.
Diagram 1: Analytical Method Transfer Workflow.
The Scientist's Toolkit: Essential Reagents and Materials
A successful method transfer relies on the careful selection and control of critical materials. The following toolkit details essential items and their functions.
Table 2: Key Research Reagent Solutions for Method Transfer
| Item | Function & Importance | Key Considerations |
|---|---|---|
| Method Transfer Kit (MTK) [107] | A centrally-managed kit containing representative, homogeneous batch(es) of material used for all transfers. | Ensures sample consistency across multiple transfers and over time; simplifies logistics. Contains pre-defined protocols. |
| Qualified Reference Standards [102] [103] | Provides the benchmark for quantifying the analyte and establishing system suitability. | Must be traceable, properly qualified, and of known purity and stability. |
| System Suitability Mixtures [106] [107] | A test preparation used to verify that the chromatographic system (or other instrument) is performing adequately. | Often contains the analyte and key impurities; critical for demonstrating method specificity and performance before sample analysis. |
| Spiked/Impurity-Enriched Samples [106] [103] | Samples where known impurities are added to challenge method accuracy, specificity, and quantitation limit at the RL. | Essential for impurity methods; proves the RL can accurately detect and quantify trace components. |
| Critical Chromatographic Reagents [102] [100] | Mobile phase components, specific columns, and buffers. | Small variations can significantly impact results (robustness). The protocol should specify suppliers and grades. Different column lots should be evaluated. |
Establishing a Framework for Ruggedness and Ensuring Data Integrity
A Proactive Approach: Building Ruggedness into Methods
Ruggedness should not be verified only at the point of transfer but should be built into the method during its development phase [100]. A systematic assessment involves:
- Identifying Critical Method Parameters (CMPs): These are the variables in the analytical procedure that, if slightly varied, could significantly impact the results. For a chromatographic method, this typically includes factors like mobile phase pH (±0.2 units), composition (±2-5%), column temperature (±2-5°C), flow rate (±10-20%), and detection wavelength [100].
- Conducting a Robustness Study: A deliberate, systematic evaluation where these CMPs are varied within a realistic, small range to mimic inter-laboratory differences. The impact on method performance (e.g., resolution, tailing factor, assay value) is measured.
- Documenting Tolerances: The established acceptable ranges for each CMP are documented in the method procedure. This provides clear guidance to the RL and is direct evidence of the method's ruggedness [101].
The Critical Role of Documentation and Communication
A method transfer is a documented process, and its success heavily depends on rigorous documentation and open communication [102] [105].
- The Transfer Protocol: This is the master document that defines the entire study. It must include the objective, scope, responsibilities of TL and RL, detailed experimental design, pre-defined acceptance criteria for all tests, and a plan for statistical evaluation [103] [104].
- The Transfer Report: This document summarizes all results, includes a statistical comparison, documents any deviations, and provides a definitive conclusion on the success or failure of the transfer [102] [105].
- Knowledge Transfer: Beyond raw data, the TL must effectively convey tacit knowledge—troubleshooting tips, historical issues, and "tricks of the trade"—to the RL [105]. This is often achieved through direct training, kick-off meetings, and collaborative problem-solving, ensuring the RL not only can perform the method but also understands it [102] [101].
In the framework of analytical method validation, the journey from establishing specificity under controlled conditions to demonstrating inter-laboratory ruggedness is critical for ensuring drug product quality globally. A successful method transfer is not an isolated event but the culmination of a well-designed, robust method and a meticulously executed, collaborative process. By adopting a lifecycle approach—incorporating ruggedness testing early in development, selecting a risk-based transfer strategy, and prioritizing clear communication and comprehensive documentation—organizations can ensure that their analytical methods consistently produce reliable and equivalent results, thereby safeguarding patient safety and upholding regulatory compliance across all manufacturing and testing sites.
Conclusion
A clear and practical understanding of specificity and selectivity is fundamental to developing robust, reliable, and regulatory-compliant analytical methods. While specificity ensures an method can accurately measure a single analyte amidst potential interferences, selectivity confirms its ability to distinguish and quantify multiple analytes within a complex mixture. Mastering these concepts, from foundational definitions through troubleshooting and final validation, directly enhances data integrity in pharmaceutical and clinical research. As analytical technologies evolve, a principled approach to specificity and selectivity will remain critical for accurately characterizing drug substances, ensuring product safety, and accelerating the development of new therapeutics.
References
- 1. What is the difference between specificity and selectivity? [https://mpl.loesungsfabrik.de/en/english-blog/method-validation/specificity-selectivity]
- 2. The Importance of the Sample Matrix [https://www.chromatographyonline.com/view/importance-sample-matrix-0]
- 3. The 6 Key Aspects of Analytical Method Validation [https://www.elementlabsolutions.com/uk/chromatography-blog/post/6-key-aspects-of-analytical-method-validation]
- 4. UNEQUIVOCAL Definition & Meaning [https://www.dictionary.com/browse/unequivocal]
- 5. Generating selectivity in analytical chemistry to reach the ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993697001222]
- 6. Key Parameters for Analytical Method Validation [https://altabrisagroup.com/analytical-method-validation-parameters/]
- 7. Introduction to Analytical Method Development and ... [https://www.labmanager.com/introduction-to-analytical-method-development-and-validation-33922]
- 8. Specificity vs Selectivity in Method Validation [https://www.linkedin.com/posts/sagar-mokashi-b225a0132_ichq2r2-usp-activity-7306337488267018240-2pkA]
- 9. Development and validation of a green/blue UHPLC-MS ... [https://www.nature.com/articles/s41598-025-15614-4]
- 10. Validation of Analytical Methods: A Review [https://www.gavinpublishers.com/article/view/validation-of-analytical-methods-a-review]
- 11. Difference between Specificity & Selectivity [https://www.linkedin.com/pulse/difference-between-specificity-selectivity-kanchan-soni]
- 12. A Guide to Analytical Method Validation [https://scioninstruments.com/us/blog/a-guide-to-analytical-method-validation-2/]
- 13. Comparing Method Validation Guidelines: What You Need ... [https://www.linkedin.com/pulse/comparing-method-validation-guidelines-what-you-need-know-pharcell-z5zrf]
- 14. FDA vs EMA: Key Differences in GMP Regulations Every ... [https://www.labmanager.com/fda-vs-ema-key-differences-in-gmp-regulations-every-pharma-lab-must-know-34062]
- 15. ( ICH ) Validation of Analytical Procedures: Text and Methodology... Q 2 R 1 [https://www.gmp-compliance.org/guidelines/gmp-guideline/ich-q2r1-validation-of-analytical-procedures-text-and-methodology]
- 16. Bioanalytical method validation - Scientific guideline [https://www.ema.europa.eu/en/bioanalytical-method-validation-scientific-guideline]
- 17. FDA vs EMA: Key Regulatory Differences for ... [https://lsacademy.com/en/fda-vs-ema-key-regulatory-differences-for-pharmaceuticals/]
- 18. Bioanalytical method : Practical approaches validation [https://www.linkedin.com/pulse/bioanalytical-method-validation-practical-approaches-zineb-chellouai-rzh0e]
- 19. Compendium of Terminology in Analytical Chemistry [https://iupac.org/compendium-of-terminology-in-analytical-chemistry-2023/]
- 20. IUPAC nomenclature of chemistry [https://en.wikipedia.org/wiki/IUPAC_nomenclature_of_chemistry]
- 21. ICH and FDA Guidelines for Analytical Method Validation [https://www.labmanager.com/ich-and-fda-guidelines-for-analytical-method-validation-34252]
- 22. Method Validation Guidelines [https://www.biopharminternational.com/view/method-validation-guidelines]
- 23. Why is Biomarker Assay Validation Different from that of ... [https://link.springer.com/article/10.1208/s12248-025-01135-5]
- 24. Analytical Method Validation: Back to Basics, Part II [https://www.chromatographyonline.com/view/analytical-method-validation-back-basics-part-ii]
- 25. Essentials in Bioassay Development [https://www.biopharminternational.com/view/essentials-bioassay-development]
- 26. Inference and design of antibody specificity [https://pmc.ncbi.nlm.nih.gov/articles/PMC11501025/]
- 27. Biological Assay Qualification Using Design of Experiments [https://www.bioprocessintl.com/assays/biological-assay-qualification-using-design-of-experiments]
- 28. Enhanced selectivity in detecting matrix metalloproteinase ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400525003612]
- 29. Advances in CE-MS Coupling for Proteomics and ... [https://www.labx.com/resources/advances-in-ce-ms-coupling-for-proteomics-and-metabolomics/5647]
- 30. Nanobore RPHPLC: Determining the Role of Selectivity in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2918168/]
- 31. A review on specific and sensitive analytical method ... [https://ijpca.org/archive/volume/10/issue/1/article/22024]
- 32. Analytical Method Validation: A Comprehensive Review ... [http://www.seejph.com/index.php/seejph/article/view/6516]
- 33. Development of forced degradation and stability indicating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5761119/]
- 34. Forced Degradation Studies: Regulatory Considerations ... [https://www.biopharminternational.com/view/forced-degradation-studies-regulatory-considerations-and-implementation]
- 35. A practical guide to forced degradation and stability studies ... [https://onyxipca.com/news/a-practical-guide-to-forced-degradation-and-stability-studies-for-drug-substances/]
- 36. Forced Degradation Studies for Biopharmaceuticals [https://www.biopharminternational.com/view/forced-degradation-studies-biopharmaceuticals-1]
- 37. What are Forced Degradation Studies? An Introduction [https://www.acdlabs.com/blog/what-are-forced-degradation-studies-an-introduction-to-pharmaceutical-stress-testing/]
- 38. AQbD-enhanced green RP-UPLC-PDA methodology for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11571561/]
- 39. Comparative Forced Degradation Study of Anticomplement ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12030638/]
- 40. An Industry Perspective on the use of Forced Degradation ... [https://www.sciencedirect.com/science/article/abs/pii/S0022354923005373]
- 41. Understanding Specificity vs Selectivity in Analytical ... [https://www.linkedin.com/posts/uttam-chand-87b69926b_analyticalmethodvslidation-regulatory-activity-7393824442247802880-f4qV]
- 42. A Look at Matrix Effects [https://www.chromatographyonline.com/view/look-matrix-effects]
- 43. Chromatography Fundamentals, Part VIII: The Meaning ... [https://www.chromatographyonline.com/view/chromatography-fundamentals-part-viii-meaning-and-significance-chromatographic-resolution]
- 44. 26.4: Optimization and Column Performance [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Instrumental_Analysis_(LibreTexts)/26%3A_Introduction_to_Chromatographic_Separations/26.04%3A_Optimization_and_Column_Performance]
- 45. Chromatographic Theory, Part V [https://www.cannabissciencetech.com/view/chromatographic-theory-part-v-chromatographic-resolution]
- 46. baseline resolution [http://www.chromforum.org/viewtopic.php?t=21110]
- 47. Methods for Changing Peak Resolution in HPLC [https://www.chromatographyonline.com/view/methods-changing-peak-resolution-hplc-advantages-and-limitations]
- 48. How to Improve HPLC Resolution: Key Factors for Better ... [https://www.mastelf.com/how-to-improve-hplc-resolution-key-factors-for-better-separation/]
- 49. Real Solutions to Improve Your HPLC Peak Resolution [https://www.thermofisher.com/blog/analyteguru/real-solutions-to-improve-your-hplc-peak-resolution/]
- 50. Validation of Stability-Indicating HPLC Methods for ... [https://www.chromatographyonline.com/view/validation-of-stability-indicating-hplc-methods-for-pharmaceuticals-overview-methodologies-and-case-studies]
- 51. HPLC and LC–MS/MS-Based Quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10856746/]
- 52. HPLC vs. LCMS: Which to Choose? 5 Key Points to Consider [https://www.hplcvials.com/news/hplc-vs-lcms-which-to-choose-5-key-points-to-consider.html]
- 53. Basic Principles of LC, HPLC, MS, & MS [https://chemyx.com/resources/knowledge-base/general-syringe-pump-info/basic-principles-hplc-ms-lc-ms/?srsltid=AfmBOop0jPRejKySBSGjZJ-WSaGQ-88h1Myudn0wLmhFHgtRDY9iugGs]
- 54. Recent Advances in Liquid Chromatography–Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204688/]
- 55. Development and Validation of an LC–MS/MS Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8742608/]
- 56. Development and validation of a highly sensitive HPLC ... [https://www.nature.com/articles/s41598-025-94907-0]
- 57. A validated LC–MS/MS method for clinical ... [https://www.nature.com/articles/s41598-023-28612-1]
- 58. Development and validation of a stability-indicating HPLC ... [https://www.sciencedirect.com/science/article/pii/S1046202324002184]
- 59. Development and Validation of an HPLC Method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6431496/]
- 60. Interference Testing and Mitigation in LC-MS/MS Assays [https://myadlm.org/cln/articles/2017/august/interference-testing-and-mitigation-in-liquid-chromatography-tandem-mass-spectrometry-assays]
- 61. Overview of Methods and Considerations for Handling ... [https://www.chromatographyonline.com/view/overview-methods-and-considerations-handling-complex-samples]
- 62. Characterization of drug interferences caused by coelution ... [https://pubmed.ncbi.nlm.nih.gov/8313597/]
- 63. Understanding Specificity and Selectivity in Analytical ... [https://www.linkedin.com/posts/vikas-bedi-37a42117b_amv-ich-ema-activity-7350335735092203520-KQJY]
- 64. Co-Elution: How to Detect and Fix Overlapping Peaks. [https://axionlabs.com/chromatography-training/co-elution-in-chromatography/]
- 65. Separation of Chromatographic Co-Eluted Compounds by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8065729/]
- 66. How to Identify Peaks in a Chromatogram, Assign Correct ... [https://www.sorbtech.com/2025/07/how-to-identify-peaks-in-a-chromatogram/]
- 67. Principles of Chromatography Method Development [https://www.intechopen.com/chapters/69775]
- 68. HPLC 2025 Preview: Boosting the Separation Power of LC ... [https://www.chromatographyonline.com/view/boosting-the-separation-power-of-lc-lc]
- 69. HPLC Method Development Steps [https://www.thermofisher.com/us/en/home/industrial/chromatography/chromatography-learning-center/liquid-chromatography-information/hplc-method-development-steps.html]
- 70. Analytical Method Validation: Complete Guide for Intended ... [https://altabrisagroup.com/guide-to-analytical-method-validation-in-pharmaceuticals/]
- 71. Strategies for Method Development and Optimization in ... [https://www.drawellanalytical.com/strategies-for-method-development-and-optimization-in-hplc/]
- 72. Optimization of HPLC methods for the development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12105191/]
- 73. Guide to GC Column Selection and Optimizing Separations [https://www.restek.com/articles/guide-to-gc-column-selection-and-optimizing-separations?srsltid=AfmBOoqvBJiNwN0clCuBhBhQSHA5Fk0HhdDjGgbn3fddXCEIJ5QKfyZg]
- 74. HPLC Method Development and Validation for ... [https://www.pharmtech.com/view/hplc-method-development-and-validation-pharmaceutical-analysis]
- 75. Box–Behnken-Assisted Optimization of High-Performance ... [https://www.mdpi.com/2297-8739/12/9/244]
- 76. The Gradient Delay Volume, Part I: Theory [https://www.chromatographyonline.com/view/the-gradient-delay-volume-part-i-theory]
- 77. Selectivity and specificity in analytical chemistry. General ... [https://link.springer.com/article/10.1007/s002160000684]
- 78. Liquid Chromatographic Peak Purity Assessments in ... [https://www.chromatographyonline.com/view/liquid-chromatographic-peak-purity-assessments-in-forced-degradation-studies---an-industry-perspective]
- 79. Calculation of Peak Purity in HPLC - Industrial Pharmacist [https://industrialpharmacist.com/2025/01/calculation-of-peak-purity-in-hplc/]
- 80. Pharmaceutical impurities and degradation products [https://www.sciencedirect.com/science/article/abs/pii/S0731708514002076]
- 81. How Analytical Method Validation Ensures Product Quality [https://www.pharmsky.com.au/how-analytical-method-validation-ensures-product-quality/]
- 82. What are system suitability tests (SST) of analytical methods? [https://mpl.loesungsfabrik.de/en/english-blog/method-validation/sst]
- 83. System Suitability Testing: Ensuring Reliable Results [https://www.labmanager.com/system-suitability-testing-ensuring-reliable-results-34248]
- 84. System suitability Requirements for a USP HPLC Method [https://www.mtc-usa.com/kb-article/aa-03965]
- 85. EDQM publishes new FAQ on System Suitability Test (SST) [https://www.gmp-compliance.org/gmp-news/edqm-publishes-new-faq-on-system-suitability-test-sst]
- 86. Why System Suitability Tests Are Not a Substitute for ... [https://www.chromatographyonline.com/view/why-system-suitability-tests-are-not-substitute-analytical-instrument-qualification-or-calibration-0]
- 87. ICH M10 Guidelines for Bioanalysts [https://labtesting.wuxiapptec.com/2025/07/30/ich-m10-what-the-finalized-guideline-means-for-bioanalytical-scientists/]
- 88. HPLC Specificity Testing: Importance Explained [https://altabrisagroup.com/hplc-specificity-testing/]
- 89. Bioanalytical method validation: An updated review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658022/]
- 90. Highlights from FDA's Analytical Test Method Validation ... [https://www.propharmagroup.com/thought-leadership/fda-analytical-test-method-validation-guidance]
- 91. 3 Critical Parameters to Examine During HPLC Validation ... [https://www.aaps.ca/blog/3-critical-parameters-to-examine-during-hplc-validation-in-pharmaceutical-school]
- 92. The Ultimate Guide to Regulatory Submissions - [https://pharmuni.com/2025/06/02/the-ultimate-guide-to-regulatory-submissions/]
- 93. 10 Essential Tips for a Successful 510(k) Submission in 2025 [https://www.thefdagroup.com/blog/510k-submission-tips]
- 94. Faster regulatory submissions for pharma with AI [https://www.mckinsey.com/industries/life-sciences/our-insights/rewiring-pharmas-regulatory-submissions-with-ai-and-zero-based-design]
- 95. The Hidden Risks in Analytical Method Validation - [https://pharmuni.com/2025/05/16/the-hidden-risks-in-analytical-method-validation/]
- 96. Challenges in Analytical Method Development and ... [https://www.biopharminternational.com/view/challenges-analytical-method-development-and-validation]
- 97. FDA Releases Guidance on Analytical Procedures [https://www.biopharminternational.com/view/fda-releases-guidance-on-analytical-procedures]
- 98. Differentiating Selectivity Vs Specificity in Pharmacology [https://altabrisagroup.com/selectivity-vs-specificity/]
- 99. Challenges in Developing Analytical Procedures with the ... [https://www.pharmoutsourcing.com/Featured-Articles/621016-Challenges-in-Developing-Analytical-Procedures-with-the-New-ICH-Q14-Guideline/]
- 100. Analytical Method Validation [https://pharmastate.academy/analytical-method-validation/]
- 101. Best Practices for Transferring Analytical Methods [https://www.jmolner.com/post/transferring-analytical-methods]
- 102. Analytical Method Transfer: Best Practices and Guidelines [https://www.labmanager.com/analytical-method-transfer-best-practices-and-guidelines-34130]
- 103. Importance of acceptance criteria in analytical method ... [https://www.gmpsop.com/importance-of-acceptance-criteria-in-analytical-method-transfer/]
- 104. Analytical Method Transfer (USP 1224) Guideline [https://pharmabeginers.com/analytical-method-transfer-usp1224-guideline/]
- 105. Best practices for analytical method transfers [https://medfilesgroup.com/best-practices-for-analytical-method-transfers/]
- 106. Points to Consider in Quality Control Method Validation ... [https://www.bioprocessintl.com/qa-qc/points-to-consider-in-quality-control-method-validation-and-transfer]
- 107. Streamlining Method Transfer Across Global Sites [https://www.biopharminternational.com/view/streamlining-method-transfer-across-global-sites]